










0. 与该篇文章相关的前情回顾
0.1 OmicsTools的安装使用和R语言分析环境的配置教程
 https://blog.csdn.net/qq_40073899/article/details/139143993
https://blog.csdn.net/qq_40073899/article/details/1391439930.2 综合的教学视频介绍
GEO数据库挖掘分析作图全流程每晚11点在线教学直播录屏回放视频: https://www.bilibili.com/video/BV1rm42157CT/
GEO数据从下载到各种挖掘分析全流程详解: https://www.bilibili.com/video/BV1nm42157ii/
一篇今年近期发表的转录组生信分析论文复现全流程直播: https://www.bilibili.com/video/BV184421Q7pj/
1. GEO数据下载教程
1.1根据GEO的GSE数据集编号自动下载和处理GEO数据教程(必须要运行的模块,GEO数据下载要首先运行这个模块)

详细教程地址: https://zhuanlan.zhihu.com/p/708053447
下载GEO数据一定要先运行下这个模块,部分数据集不一定能下载提取出表达矩阵,但是一般能下载到非常完整的样本的注释信息等数据文件,这在对GEO数据集临床信息分析或分组差异分析是非常重要的。
当然如果少数GSE数据集遇到没有提取出GSEXXX_sample_info.csv的情况,也可以看看我b站的教学视频,根据GEO网页中的样本编号的分组情况,自建一个这样的分组文件,分组文件一般是长这个样子的,有两列,一列列名叫sample.id,是GSM编号信息,另一列是group.level,存放的是分组信息,当然sample.id这一列的样本名还是要以表达矩阵的样本名为准,这样才能让表达矩阵后面跟样本分组信息按照相同的列名整合在一起:

对于部分没有下载出表达矩阵的数据集,如果该数据集的GEO网页中有GSEXXX_RAW.tar格式的压缩包,可以运行下面的1.2这一步对RAW.tar的压缩包文件进行提取和处理。
1.2 转录组和基因芯片GSE数据集RAW.tar压缩包下载和多样本整合处理教程 (GEO网页中有RAW.tar压缩包可以用这一步下载处理)
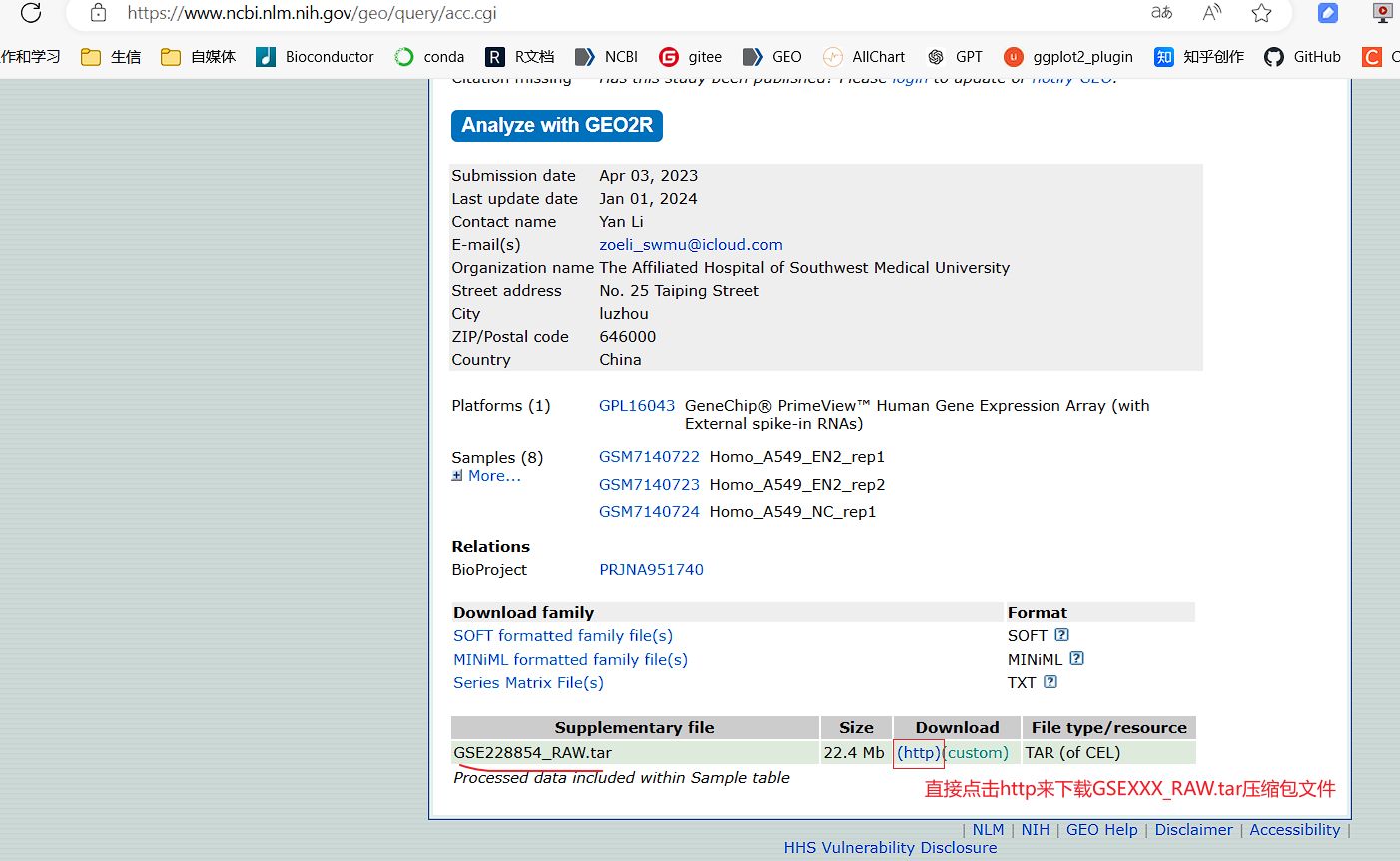
教程地址
教程地址:转录组和基因芯片GSE数据集RAW.tar压缩包下载和多样本整合处理教程 - 邢博士谈科教的文章 : https://zhuanlan.zhihu.com/p/708244032
普通RNAseq转录组的GSEXXX_RAW.tar压缩包的多样本整合处理
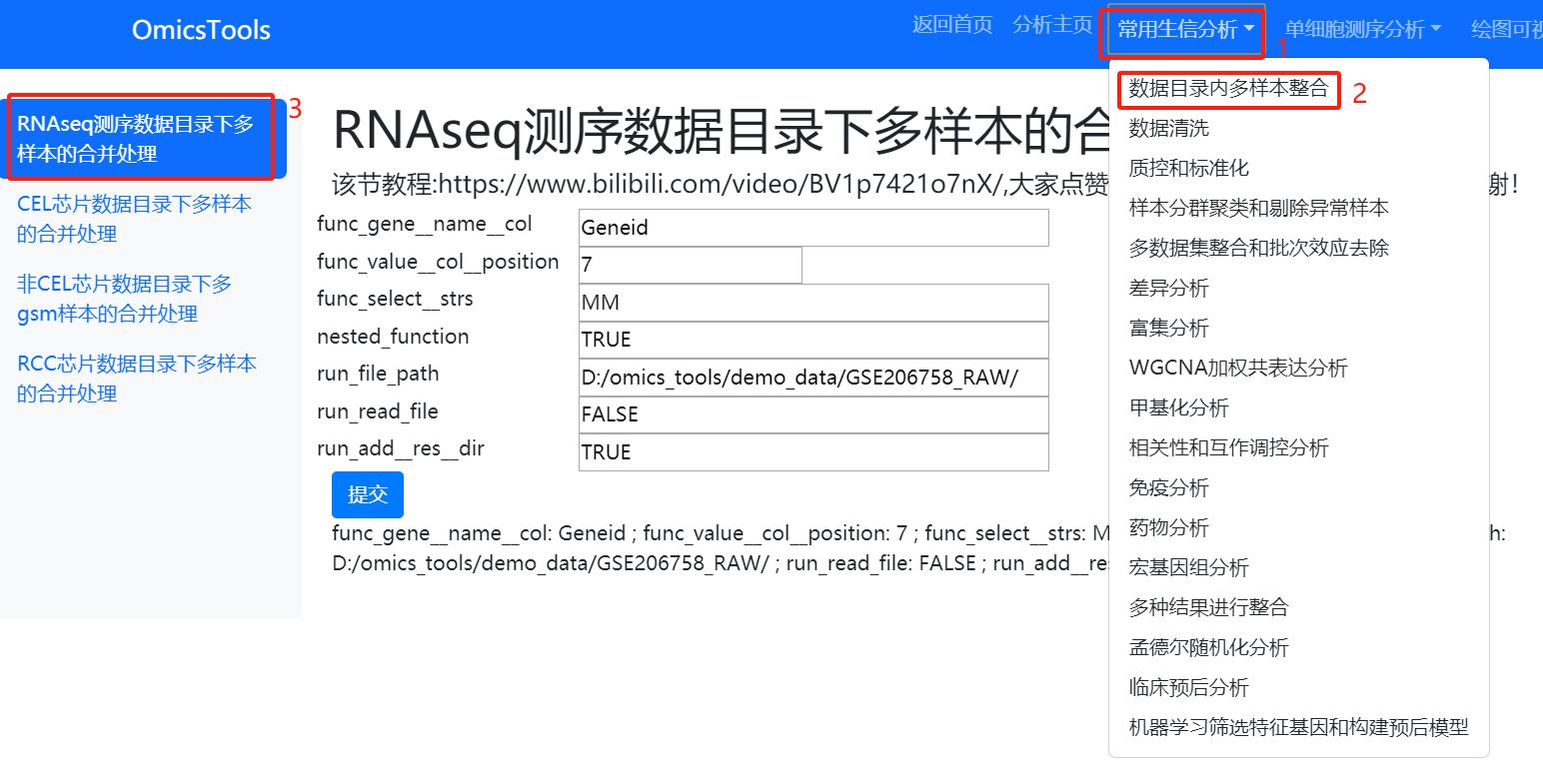
各种不同格式的基因表达芯片的GSEXXX_RAW.tar压缩包的多样本整合处理

1.3 GEO网页中存在可以手动下载的表达矩阵等文件的下载处理教程
如果在1.1步没有自动下载提取出基因表达矩阵,GEO网页中也没有GSEXXX_RAW.tar压缩包文件,但是存在可以手动下载的一些表达矩阵等数据文件,可以用这一步进行下载和处理。
一般每个 GSE数据集都有一个GPL平台信息,GPL平台信息有seq单词的是转录组测序数据,带array或chip的芯片数据,GSEXXX_RAW.tar压缩包解压后每个GSM文件的文件名后缀中有CEL字符的是CEL芯片,有RCC字符的是RCC芯片
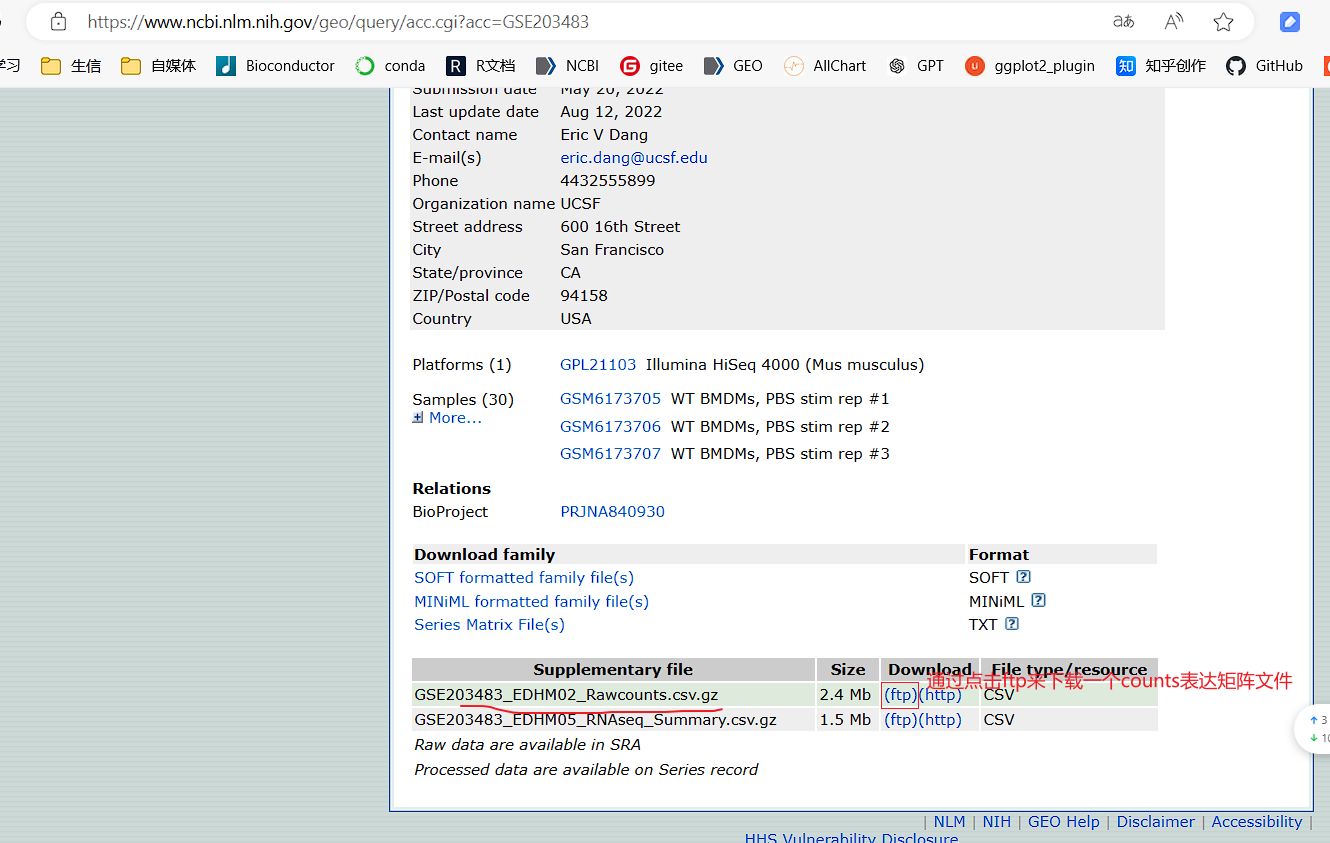

教程视频:https://www.bilibili.com/video/BV1Yf421X7Cj/
2. GEO数据处理清洗教程
2.1 GEO的表达矩阵的探针ID转换成基因名称教程 (提取的表达矩阵里只有基因探针ID,没有正式的基因名称时运行这一步)
2.1.0 教程地址
GEO的表达矩阵的探针ID转换成基因名称教程 - 邢博士谈科教的文章 - 知乎
https://zhuanlan.zhihu.com/p/708404618
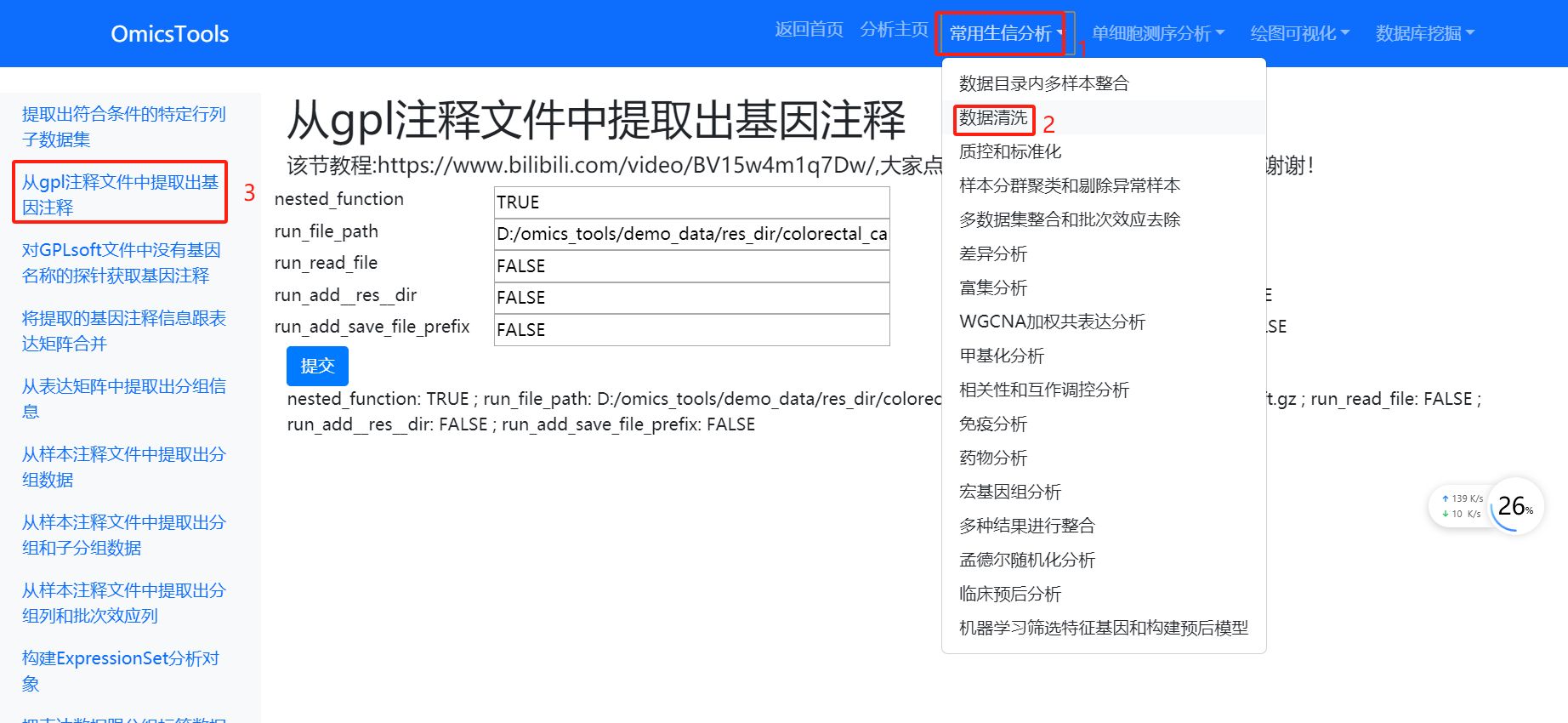
2.1.1 从gpl注释文件中提取出基因注释 (优先用这种方法进行基因名称注释)
该节教程:https://www.bilibili.com/video/BV15w4m1q7Dw/
2.1.2 对GPLsoft文件中没有基因名称的探针获取基因注释(第二种基因探针注释方法)
该方法对于一些非编码RNA的GSE数据集的注释可能会非常有用
该节教程:https://www.bilibili.com/video/BV12b421e7SX/
2.1.3 同一物种不同类型基因id转换
该节教程:https://www.bilibili.com/video/BV1YD421M7qR/
该模块对于基因名称SYMBOL跟ENTREZID,ENSEMBL(为ENSG等开头基因ID),REFSEQ(为NM开头:mRNA,NP开头:蛋白,NR开头:非编码RNA),UNIPROT,UCSCKG(为ENST转录本)这六种类型中的一中或多种类型的基因ID或名称间的互相转换是非常有用的。

2.1.4 不同物种间同源基因转换
该节教程:https://www.bilibili.com/video/BV1B1421k7K9/
该方法对于小鼠和人之间的基因名称相互转换或者其它物种的基因名称转成人或小鼠的基因名称从而方法进行某些只能用人或小鼠模式生物才能做的生信分析是非常有用的。
2.2 将提取的基因注释信息跟表达矩阵合并
该节教程:https://www.bilibili.com/video/BV12f421Q7nK/
2.3 GEO的样本分组信息提取和处理教程
2.3.1 从样本注释文件中提取出分组数据 (优先选择使用这种方法)
该节教程: https://www.bilibili.com/video/BV1rT421v77P/
2.3.2 从表达矩阵中提取出分组信息 (第2种分组信息提取方法)
该节教程:https://www.bilibili.com/video/BV1sz421h7rc/
2.3.3 自己构建分组信息文件
当然如果少数GSE数据集遇到没有提取出GSEXXX_sample_info.csv的情况,也可以看看我b站的教学视频,根据GEO网页中的样本编号的分组情况,自建一个这样的分组文件,分组文件一般是长这个样子的,有两列,一列列名叫sample.id,是GSM编号信息,另一列是group.level,存放的是分组信息,当然sample.id这一列的样本名还是要以表达矩阵的样本名为准,这样才能让表达矩阵后面跟样本分组信息按照相同的列名整合在一起:

3. 质控和标准化
3.1 基因表达芯片的质控和标准化
3.1.1构建ExpressionSet分析对象 (首先先运行这一步)
基因的芯片的质控需要的数据格式是ExpressionSet这种格式,所以先需要构建个ExpressionSet对象
该节教程:https://www.bilibili.com/video/BV1br421c71A/
3.1.2 基因表达芯片的质控
该节教程: https://www.bilibili.com/video/BV1vy411b7yQ/
3.1.3表达芯片进行归一化处理
该节教程: https://www.bilibili.com/video/BV1fJ4m1u7jX/
3.2 普通转录组RNAseq定量的表达矩阵质控和标准化的操作和答疑
3.2.0 普通转录组RNAseq定量的表达矩阵是否需要质控和标准化的答疑
RNAseq测序数据在上游处理分析时候已经经过质控处理,是不需要再进行质控的,我们一般从GEO数据库里下载处理的RNAseq测序数据的表达矩阵要么是counts整数值表达矩阵文件,要么可能是FPKM/RPKM或TPM相对定量且标准化后连续性数值表达矩阵。
在差异分析的时候,转录组的差异分析主流的DESeq2和edgeR这两个差异分析工具是需要用不经过任何处理的原始的counts表达矩阵作为输出数据的,所以我们是不需要对从GEO数据库中下载和提取的counts表达矩阵做任何质控标准化操作的。
如果我们没有counts表达矩阵,只下载到了FPKM/RPKM或TPM相对定量且标准化后连续性数值表达矩阵,这样的数据已经经过了标准化处理,在我们做差异分析的时候,只需要对该类表达矩阵数据取个log2对数处理就可以了,同时这样的数据已经不能使用DESeq2和edgeR做差异分析了,只能用limma这个工具做差异分析。而在我的OmicsTools差异分析模块用limma做差异分析时,是会自动对数据分布和量级差别较大的数据进行取对数处理的。所以,大家也不需要手动对这类数据提取进行取对数处理的。
对于FPKM/RPKM或TPM数据,如果大家在做limma差异分析之前,如果想先看下RNAseq的表达矩阵每个样本所有基因的表达数据的分布情况是否一致,可以用下面的这两个模块进行表达矩阵的可视化作图
3.2.1 使用表达矩阵进行密度图绘制
该绘图模块会绘制每个样本的表达数据的密度曲线,好的连续数据的密度曲线应该近似钟形曲线的正态分布。
视频教程:https://www.bilibili.com/video/BV1yx4y1W7Vb/

3.2.2 利用表达矩阵进行箱式图绘制
利用表达矩阵进行箱式图绘制(用于查看质控标准化结果) ,质量好的数据应该是所有样本的箱式图中的中位数应该位于同一水平线上,箱式图分布性状也比较近似,没有特别严重的高低不齐的现象。
视频教程: https://www.bilibili.com/video/BV1b1421b7Qx/
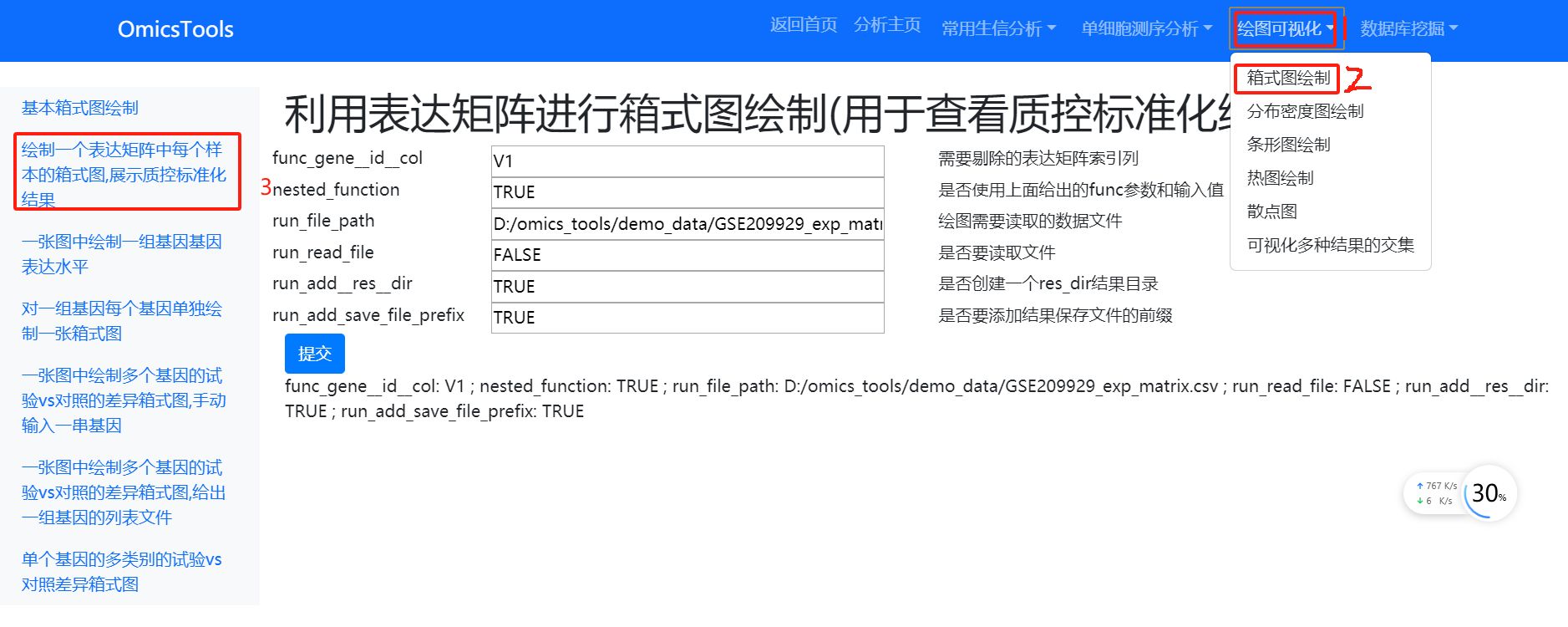
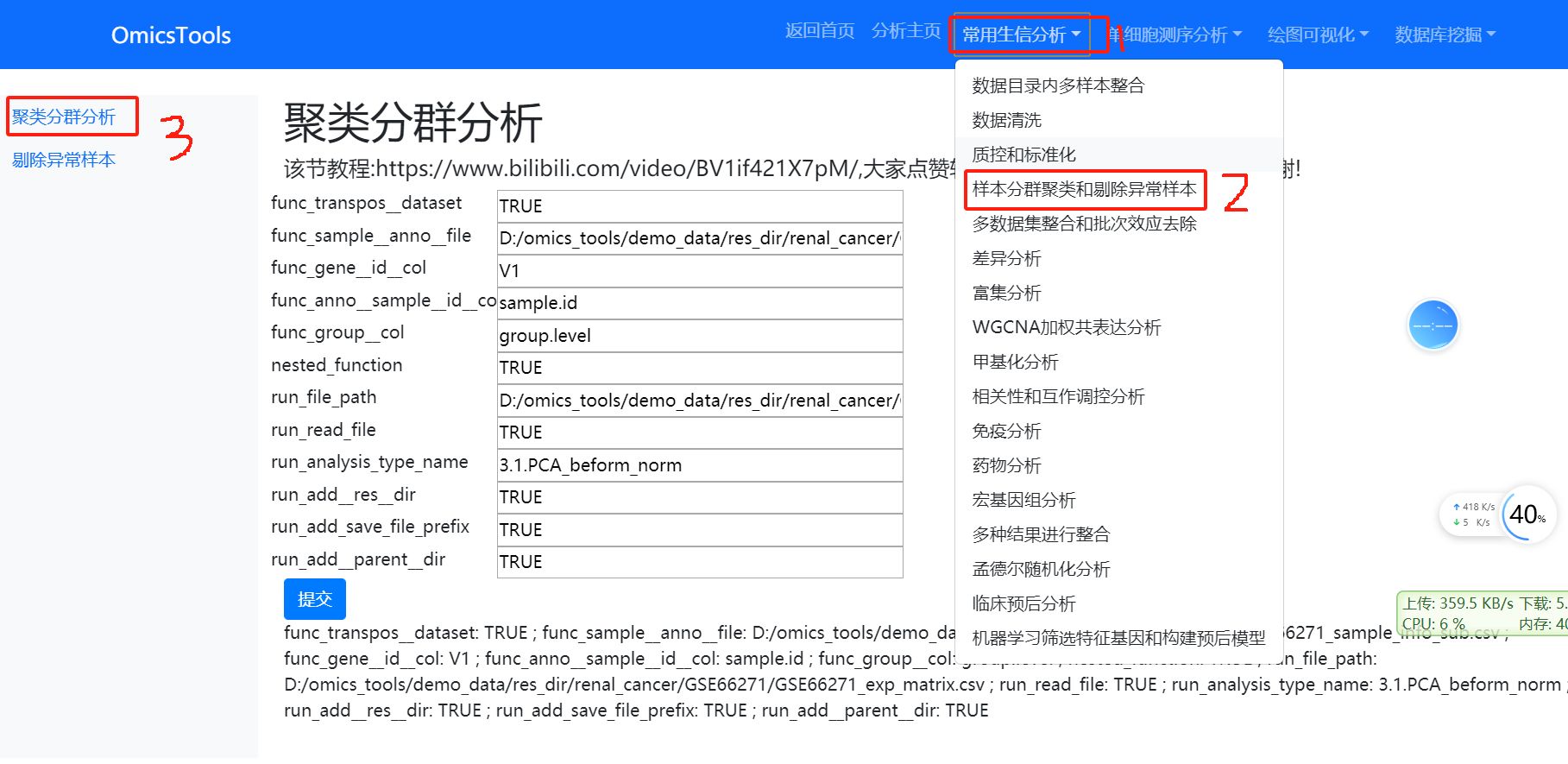
4. 样本PCA分群聚类分析和剔除异常样本
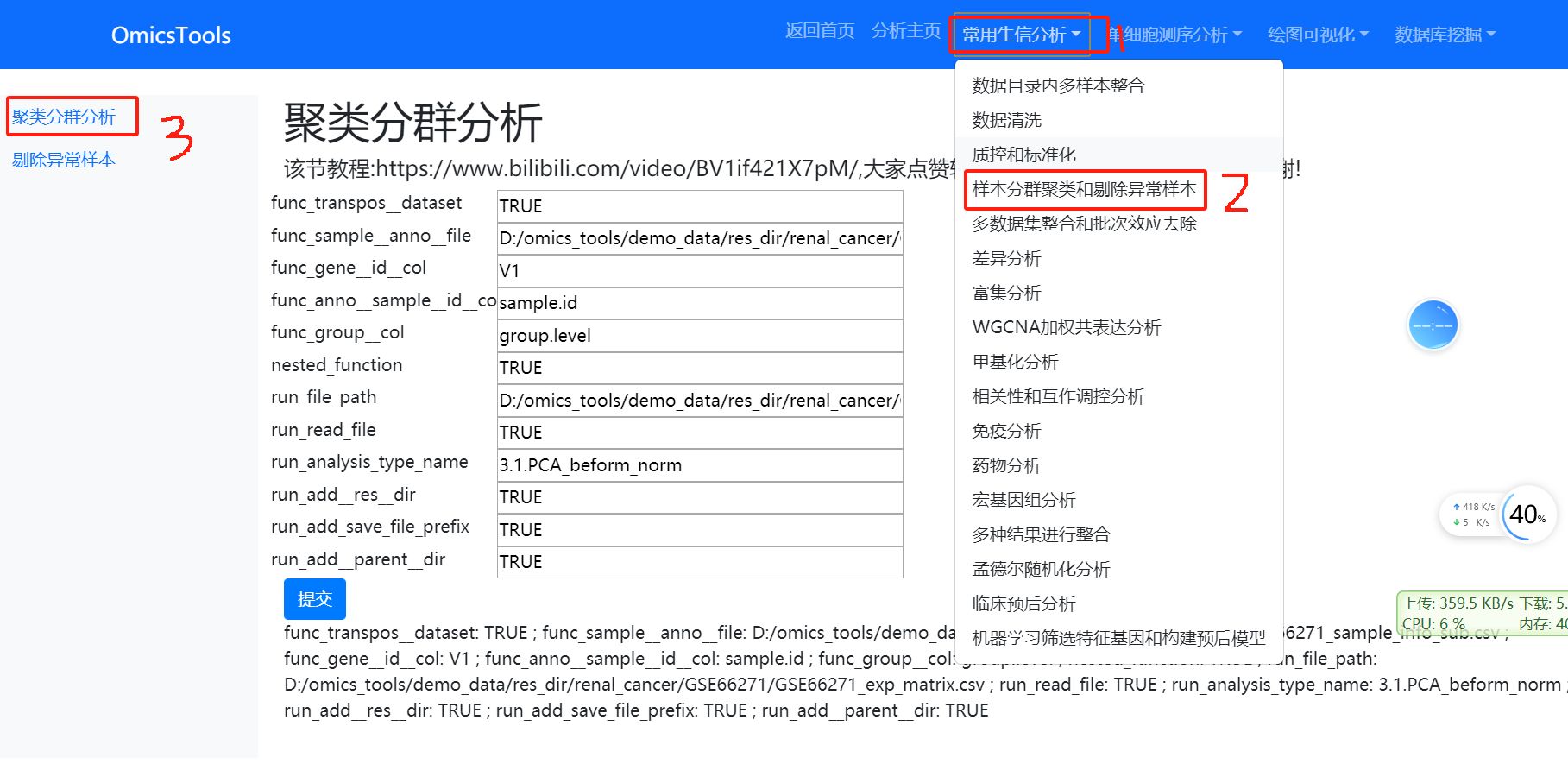
4.1 样本的PCA聚类分群分析
该节教程:https://www.bilibili.com/video/BV1if421X7pM/
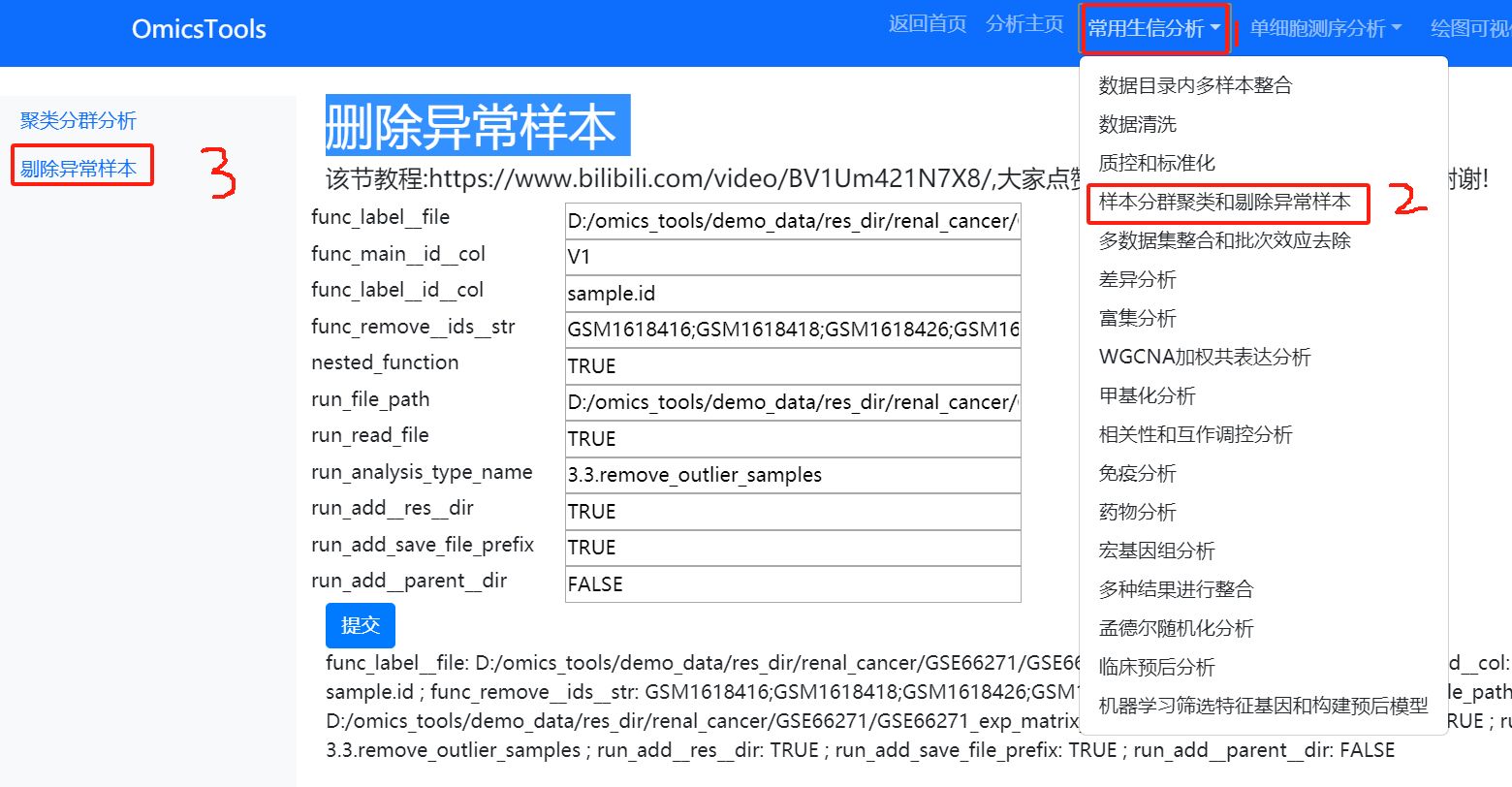
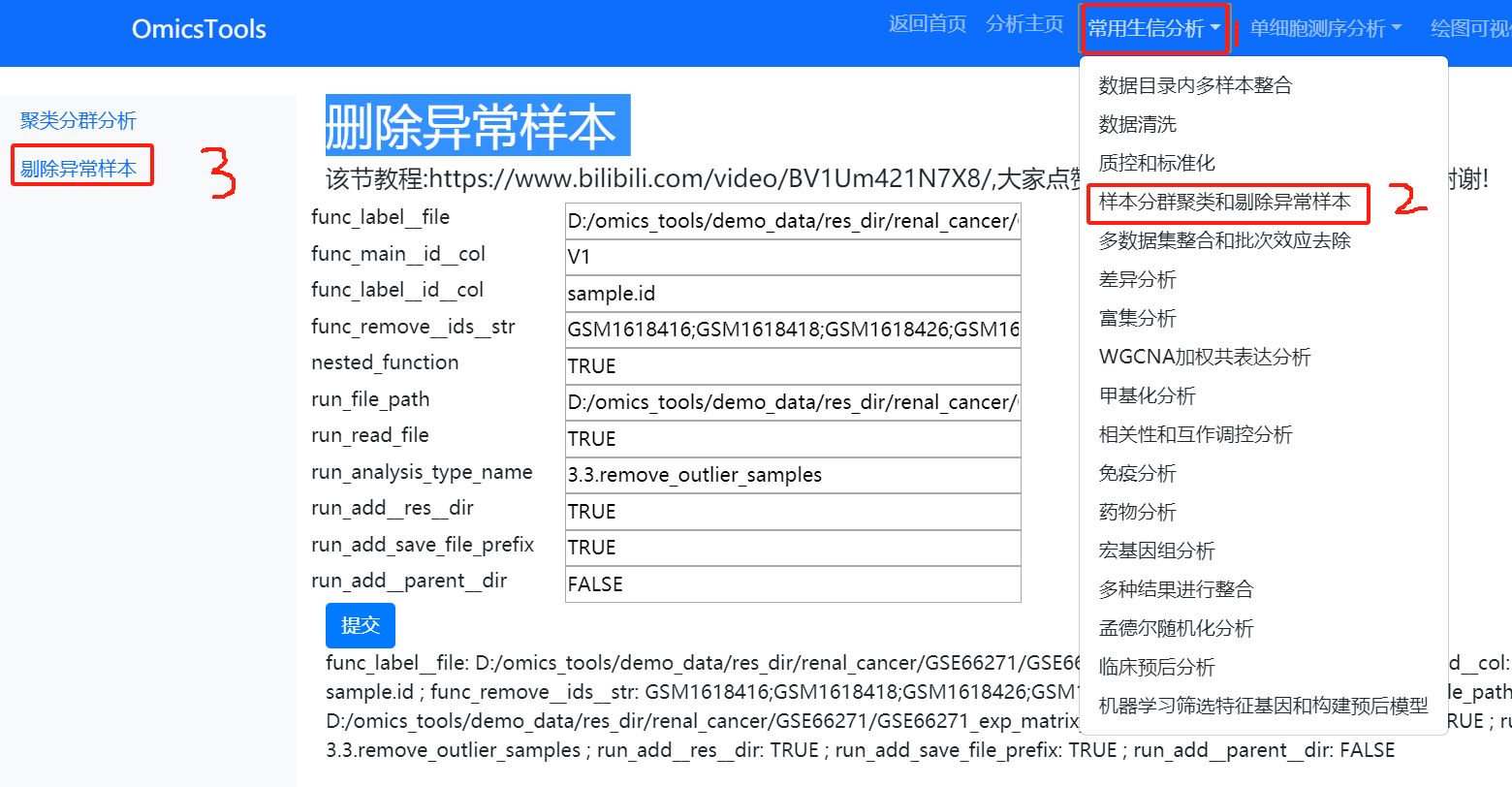
4.2 删除PCA分群聚类的异常离群样本
该节教程:https://www.bilibili.com/video/BV1Um421N7X8/
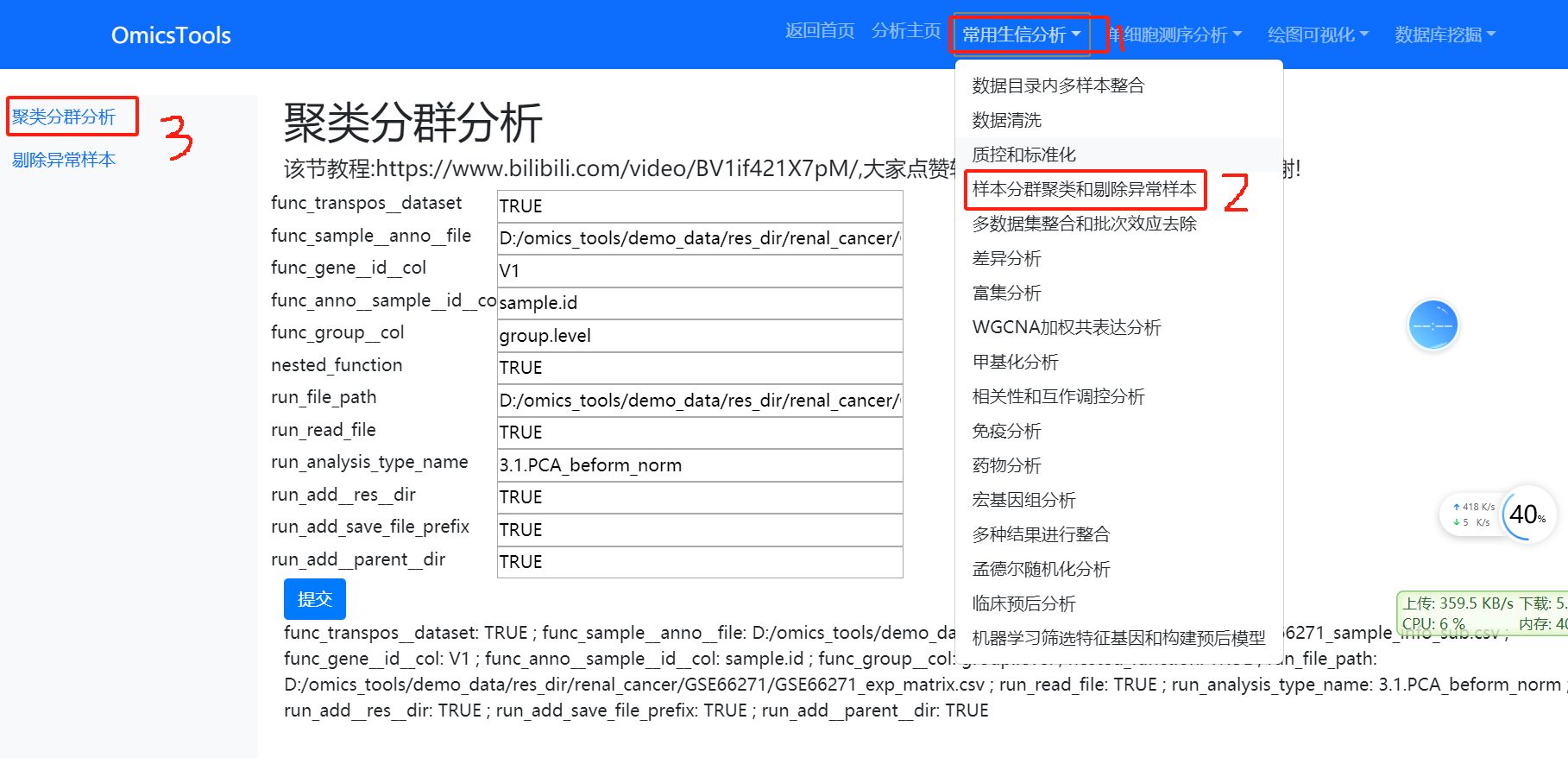
4.3 剔除异常离群样本后的再次PCA聚类分群分析
该节教程:https://www.bilibili.com/video/BV1if421X7pM/
5. 多个数据集的整合和去除批次效应
5.1 多个数据集合并 (如何需要整合分析多个数据集,就运行这一步)
该节教程:https://www.bilibili.com/video/BV1j4421X7Ls/
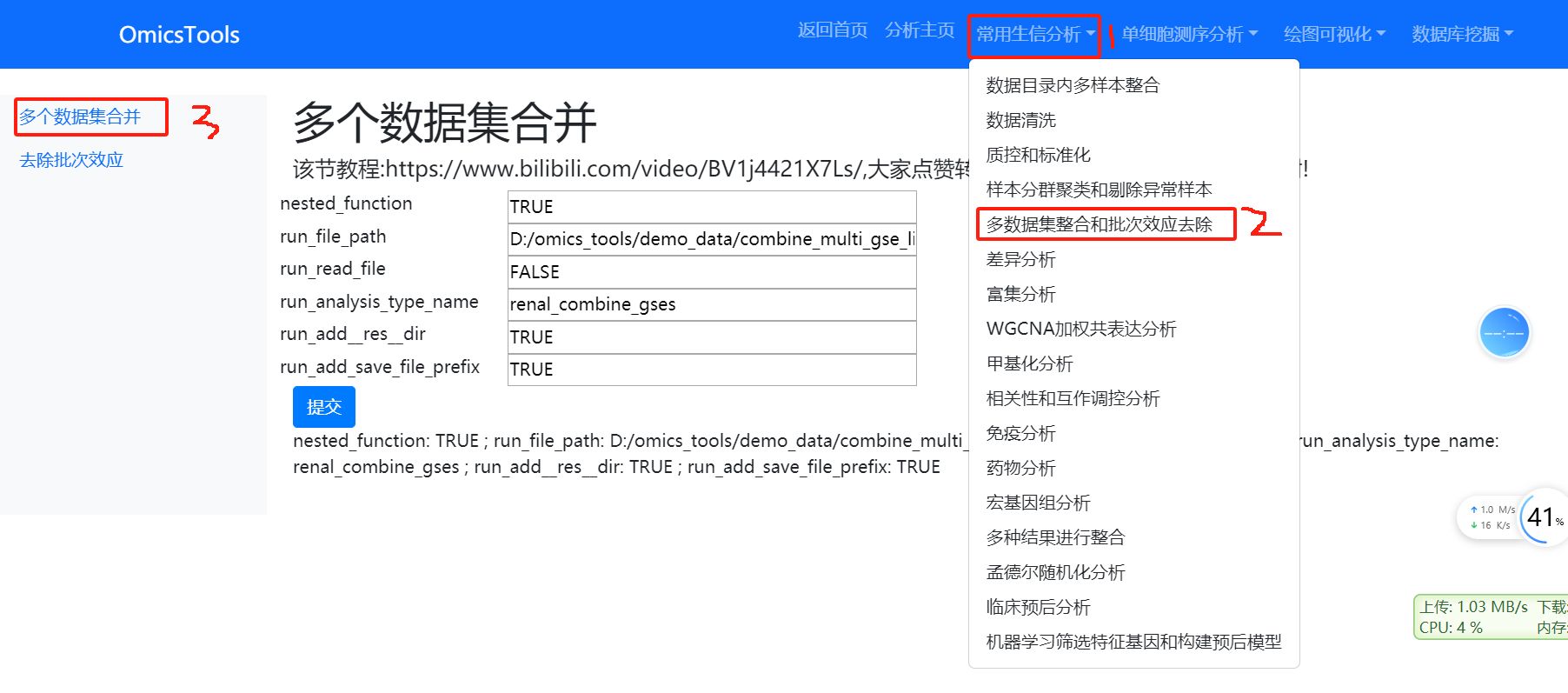
5.2 多数据集合并后一起所有样本表达数据一起做归一化处理(RNAseq counts整型数据不需要做这一步)
该节教程: https://www.bilibili.com/video/BV1fJ4m1u7jX/
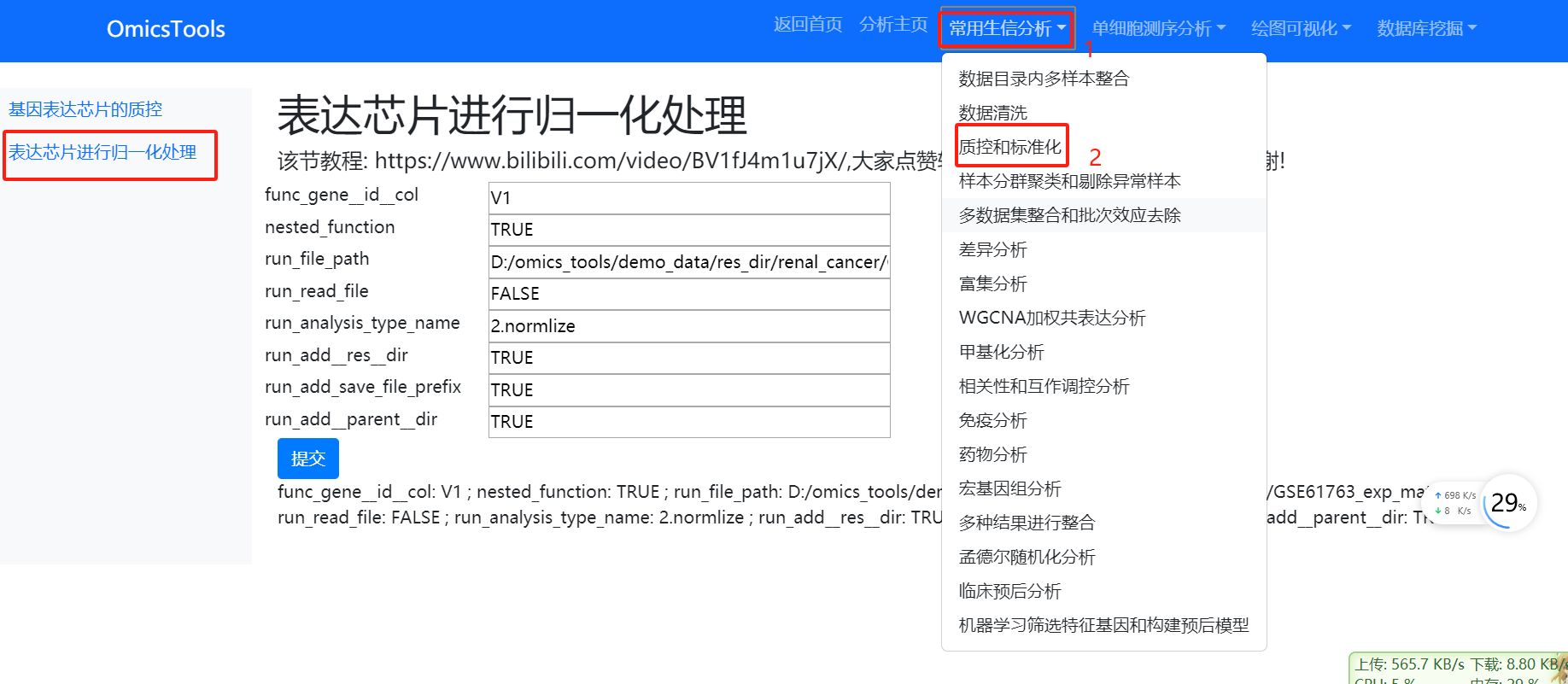
5.3去除批次效应(即使单个数据集中有多个批次信息,也要运行这一步)
一般我们如果合并了多个数据集,都是需要要去除批次效应的,但是,即使单个数据集中如果有多个批次信息,也要运行这一步
该节教程: https://www.bilibili.com/video/BV1Yy411h78f/
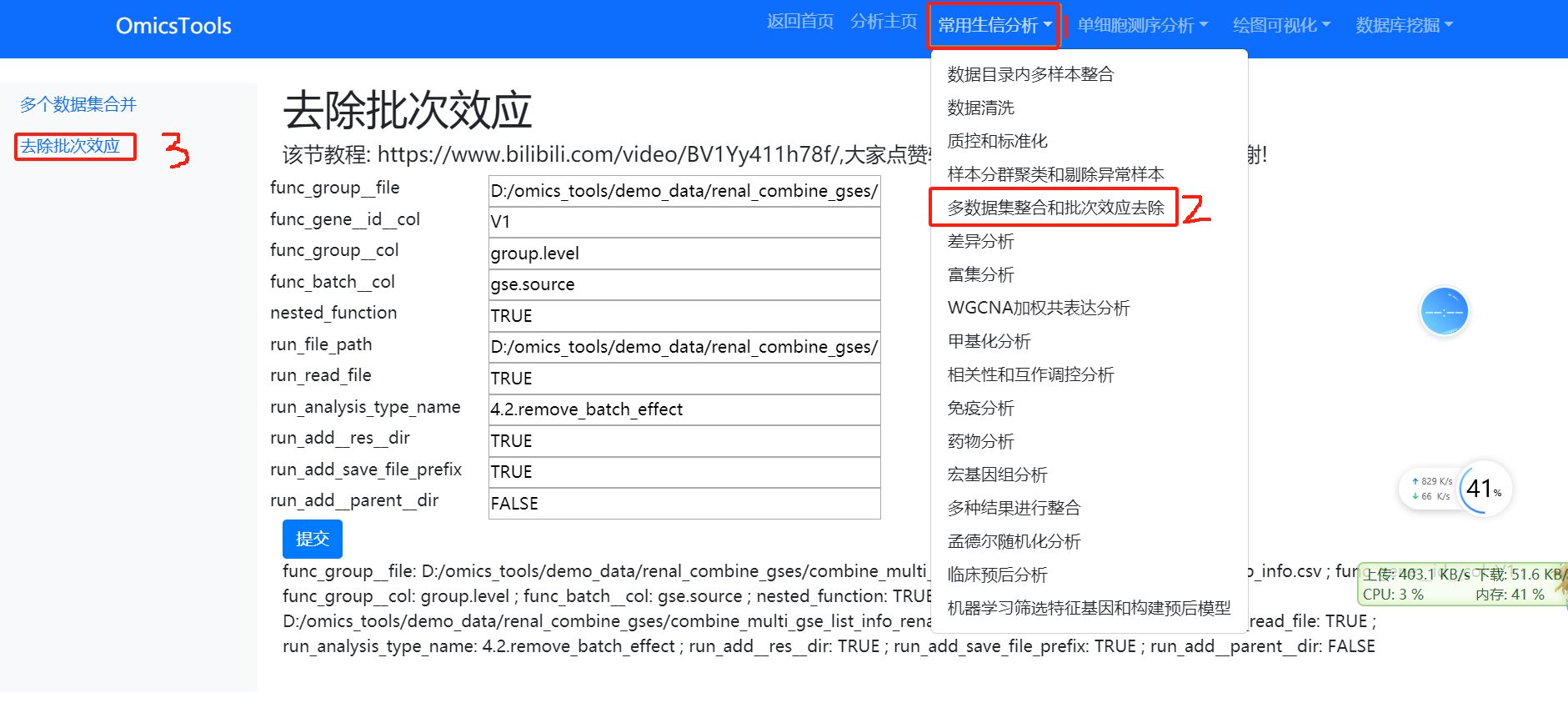
5.4 去除批次效应后的样本PCA聚类分群分析
该节教程:https://www.bilibili.com/video/BV1if421X7pM/
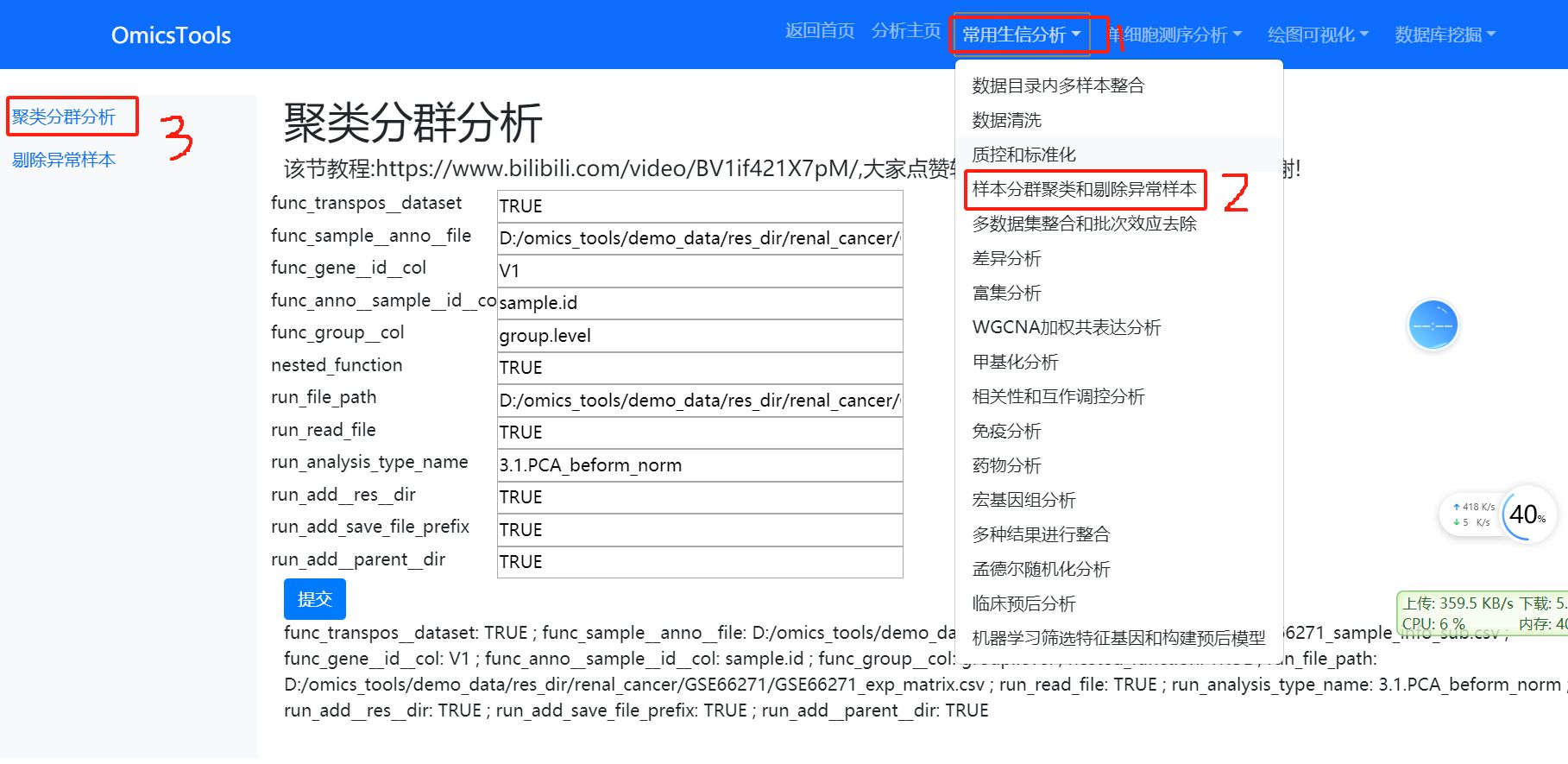
5.4 删除PCA分群聚类的异常离群样本
该节教程:https://www.bilibili.com/video/BV1Um421N7X8/
5.5 剔除异常离群样本后的再次PCA聚类分群分析
该节教程:https://www.bilibili.com/video/BV1if421X7pM/
6. 差异分析
6.0 不同数据的差异分析方法选择
6.0.1 整个转录组所有基因批量差异分析的数据格式和方法选择
数据格式: counts数据>> TPM数据> >FPKM/RPKM数据
差异分析工具: DESeq2 =edgeR > >(log2对数标准化+limma)
1.有counts数据的时候,优先选择使用counts数据+DESeq2/edgeR工具进行整个转录组的批量差异分析
2.没有counts数据,但是有TPM数据的时候,优先选择使用TPM数据+(log2对数标准化+limma工具)进行整个转录组的批量差异分析
3.没有counts数据,也没有TPM数据的时候,可以选择使用FPKM/RPKM+(log2对数标准化+limma工具)进行整个转录组的批量差异分析。
在我的OmicsTools差异分析模块用limma做差异分析时,是会自动对数据分布和量级差别较大的数据进行取对数处理的。所以,大家也不需要手动对这类数据提取进行取对数处理的。
6.0.2 基因芯片或其它组学或连续数值的表达矩阵的差异分析方面选择
基因芯片或其它组学或连续数值的表达矩阵的差异分析时,一律是log2(表达矩阵)+limma工具差异分析这种方法。在我的OmicsTools差异分析模块用limma做差异分析时,是会自动对数据分布和量级差别较大的数据进行取对数处理的。所以,大家也不需要手动对这类数据提取进行取对数处理的。
6.1 通用差异分析 (各种类型的组学数据都适用的差异分析模块)
该节教程:https://www.bilibili.com/video/BV1Lw4m1q71T/
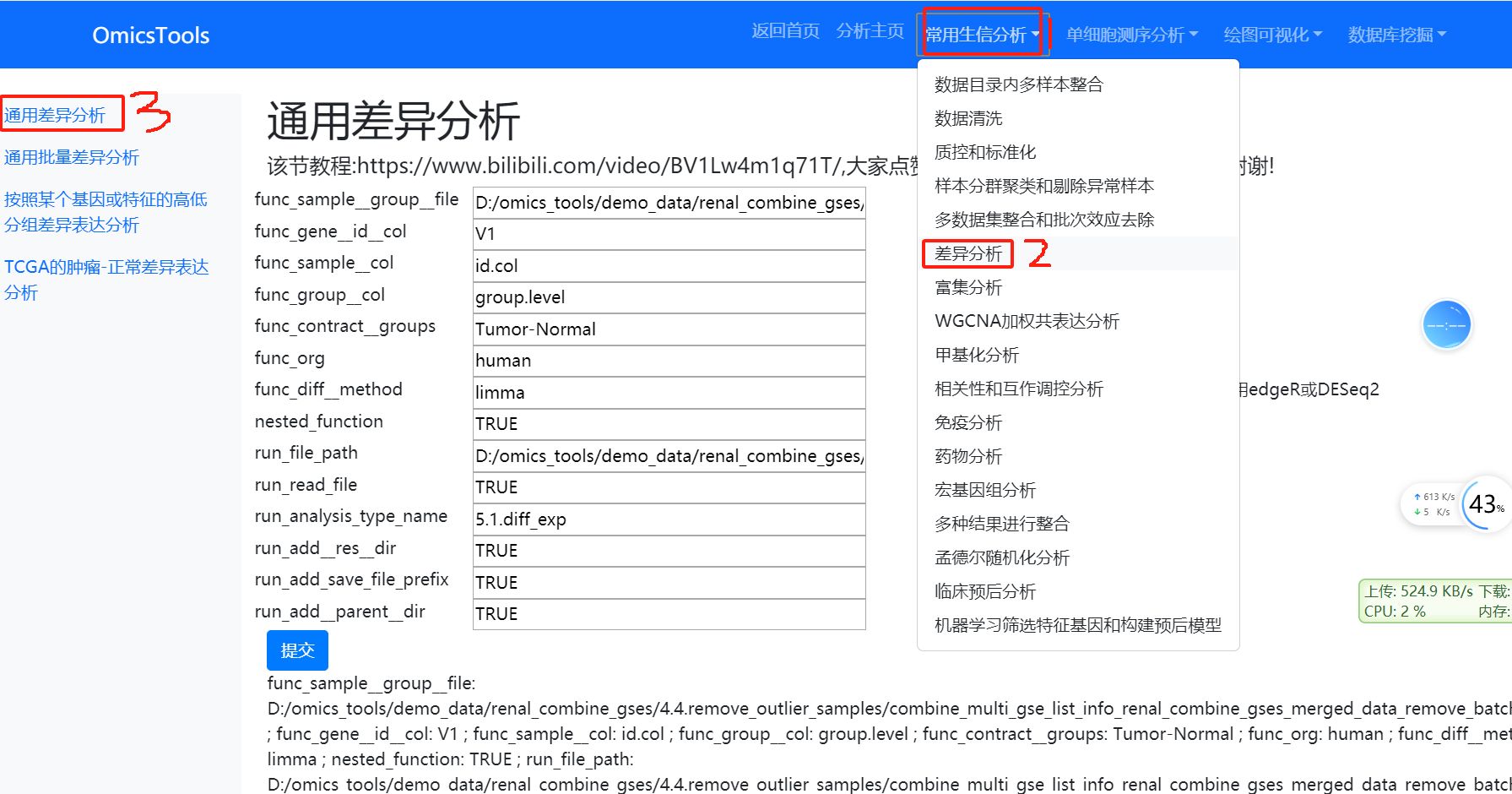
这个差异分析模块是可以做任意物种的差异分析,如果是人,func_org参数就填’human’, 如果不是人,就填不是’human’的任意其它物种名称就行。
因为edgeR差异分析建议,如果是人类的RNAseq数据,bcv生物变异度设置为0.4,如果是非人类的模式生物,bcv设置为0.1,在用edgeR做差异分析时,所有当输入的物种是human的时候,会自动把bcv生物变异度设置为0.4, 非human的时候,bcv会设置为0.1
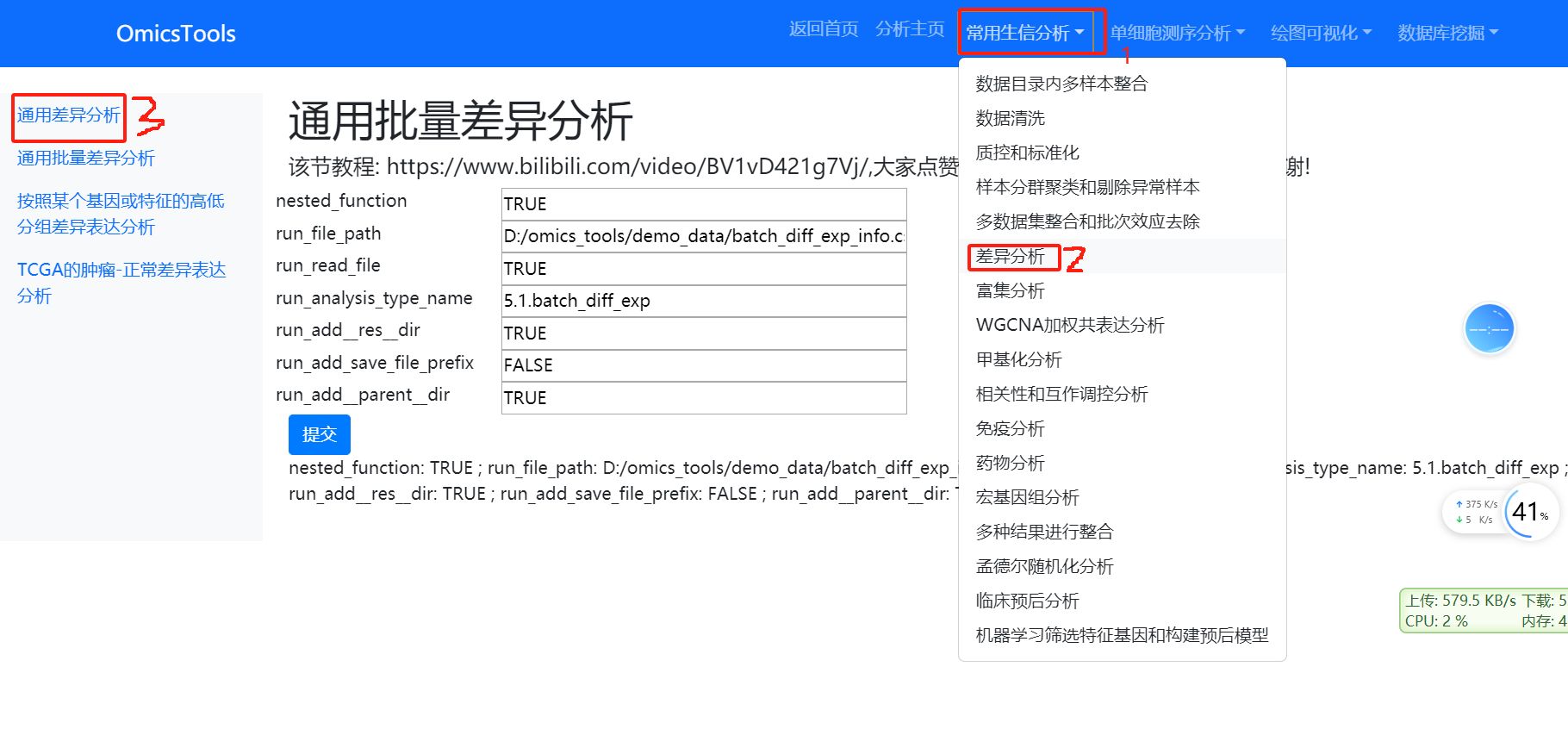
6.2 通用批量差异分析(可同时对多个数据集文件做差异分析)
该节教程: https://www.bilibili.com/video/BV1vD421g7Vj/
6.3 按照某个基因或特征的高低分组的差异分析
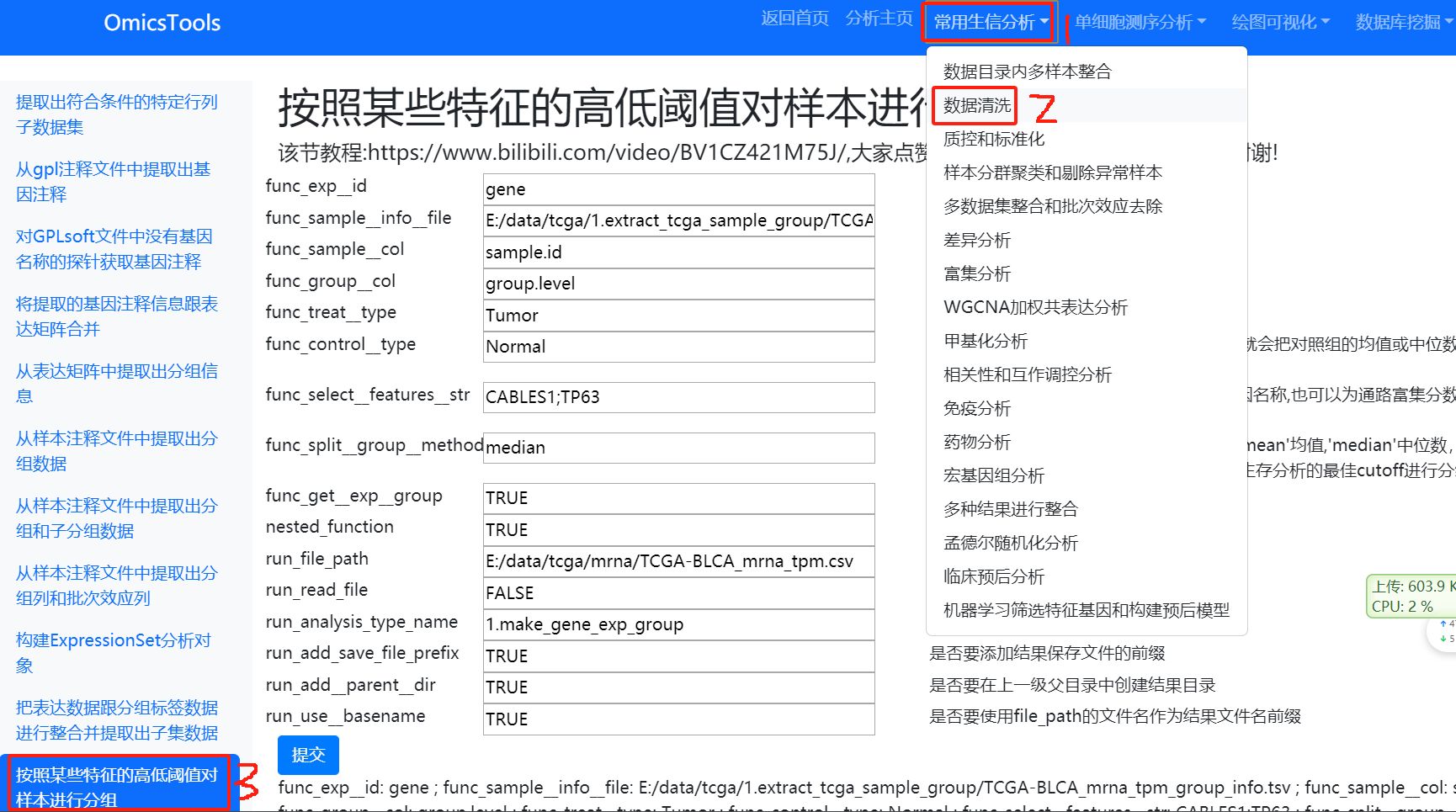
6.3.1 按照某些特征的高低阈值对样本进行分组
该节教程:https://www.bilibili.com/video/BV1CZ421M75J/
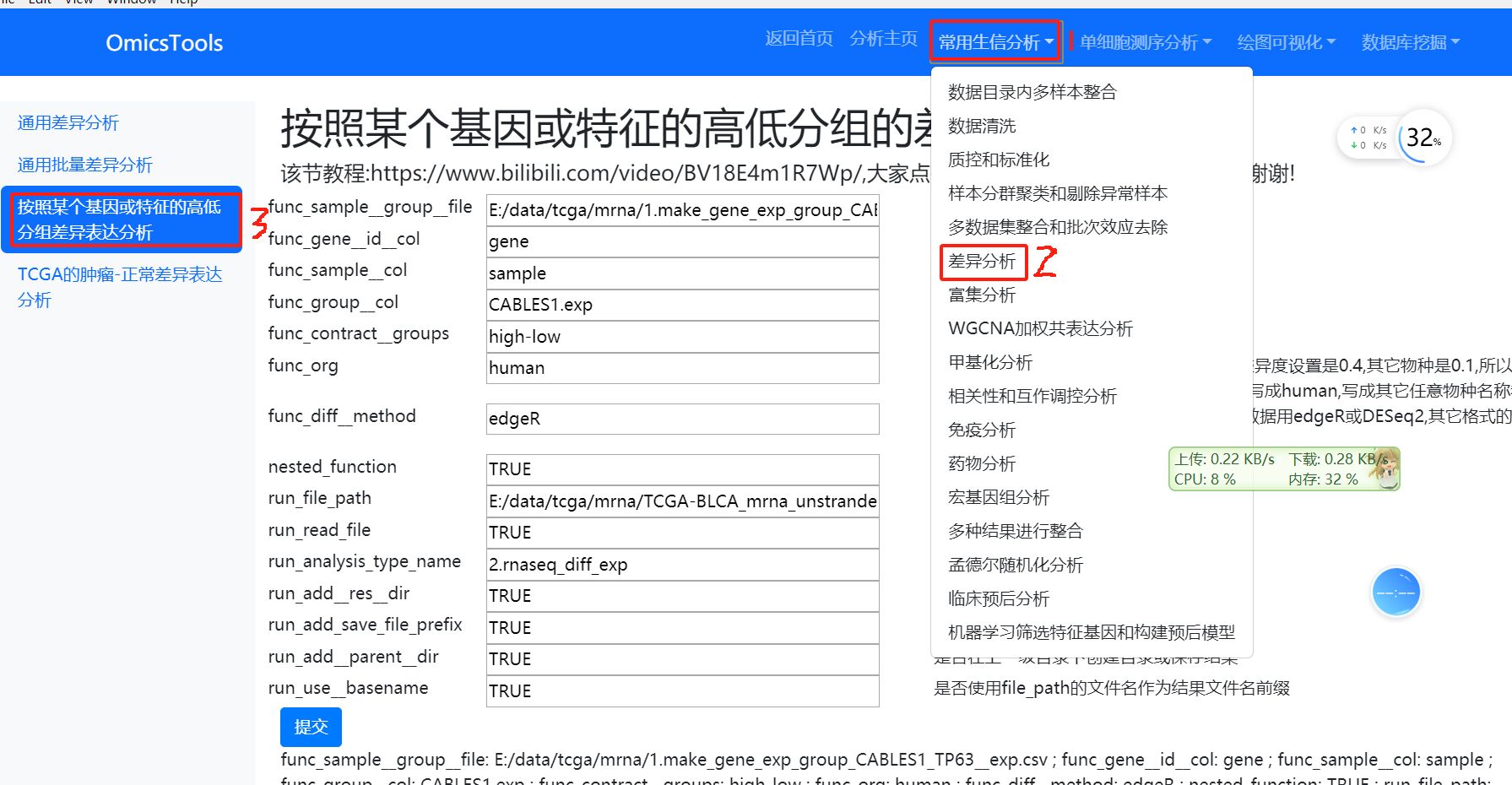
6.3.2 按照某个基因或特征的高低分组的差异分析
按照某个基因或特征的高低分组的差异分析
该节教程:https://www.bilibili.com/video/BV18E4m1R7Wp/
这个差异分析模块也是可以做任意物种的差异分析,如果是人,func_org参数就填’human’, 如果不是人,就填不是’human’的任意其它物种名称就行。
因为edgeR差异分析建议,如果是人类的RNAseq数据,bcv生物变异度设置为0.4,如果是非人类的模式生物,bcv设置为0.1,在用edgeR做差异分析时,所有当输入的物种是human的时候,会自动把bcv生物变异度设置为0.4, 非human的时候,bcv会设置为0.1
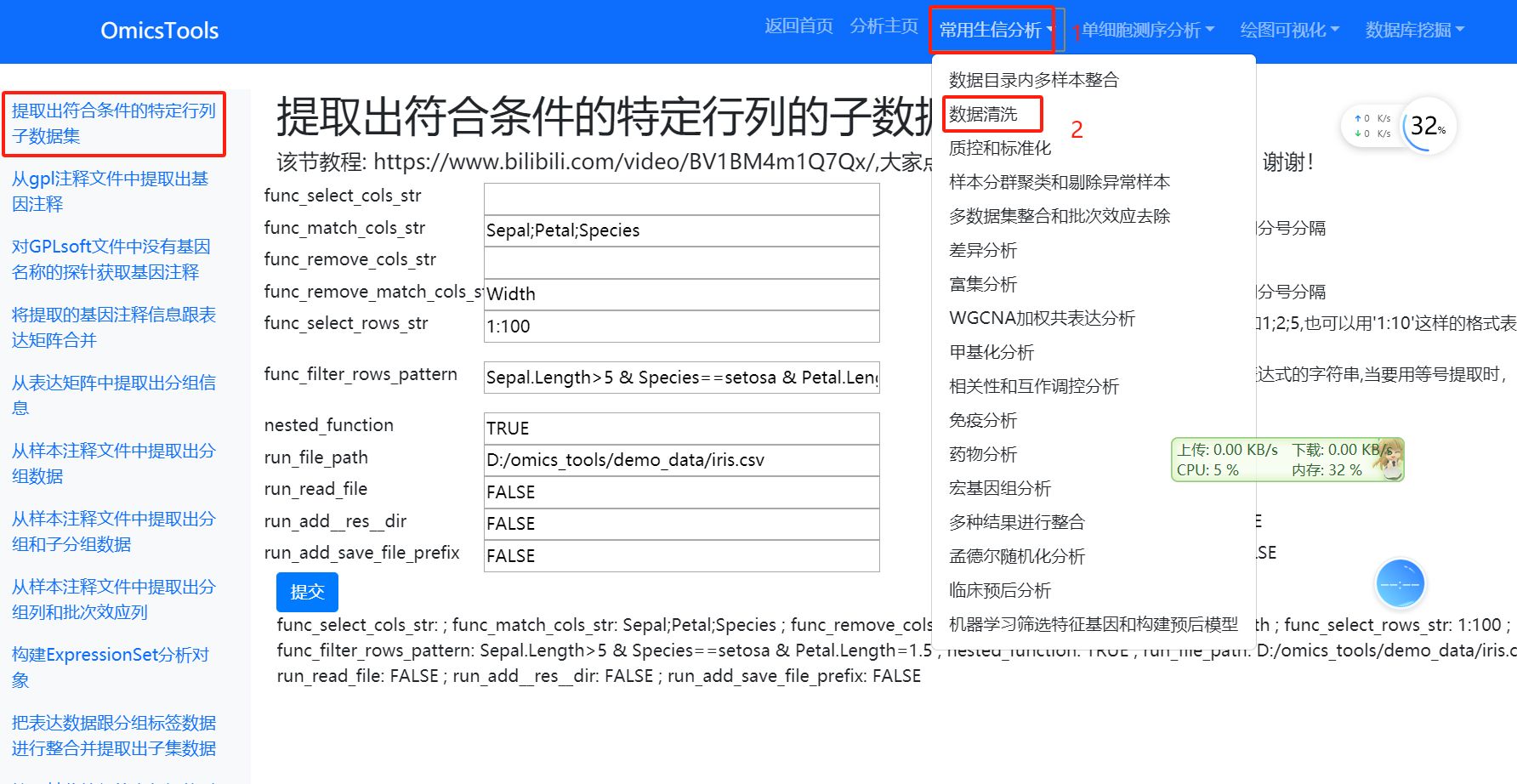
6.4 根据logFC和p值等条件对差异分析结果进行筛选
提取出符合条件的特定行列的子数据集
该节教程: https://www.bilibili.com/video/BV1BM4m1Q7Qx/


















 1826
1826

 被折叠的 条评论
为什么被折叠?
被折叠的 条评论
为什么被折叠?
 到【灌水乐园】发言
到【灌水乐园】发言











