







表型文件
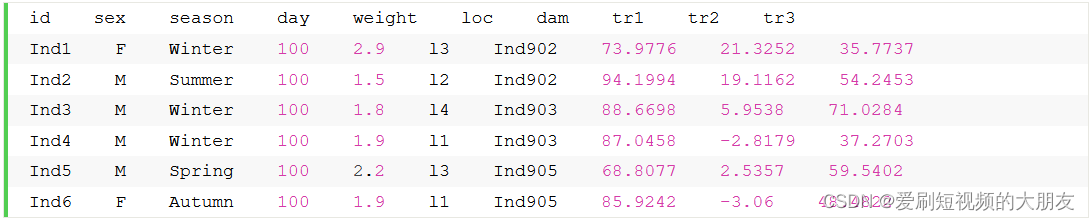
与其他软件不同,HIBLUP 所需的表型文件不仅要包括表型记录,还要包括环境协变量、固定效应和随机效应。第一列必须是单独的 id,头部应该包含在文件中。示例表型文件简介如下:
 下面的任何符号在加载文件时将被视为缺失:
下面的任何符号在加载文件时将被视为缺失:
 此文件应该分配给参数选项
此文件应该分配给参数选项--pheno。
默认情况下,HIBLUP 将采用第二列的 trait 进行分析,用户可以通过使用--pheno-pos n,例如--pheno-pos 8表示,使用第8列进行分析来指定不同的列。
注意
如果有多个性状可用于估计遗传相关性,请使用空格作为分隔符,例如--pheno-pos 8 9 10。
协变量,固定和环境随机效应
对于一个性状的协变量、固定效应和环境随机效应,请分别在表型文件中指定它的位置来标记--qcovar(协变量)、--dcovar(固定效应)、--rand(随机效应)。
对于单个 trait(性状),请使用逗号","作为分隔符,例如:
--qcovar 4,5将“day” and “weight”视为协变量。
--dcovar 2,3将“sex” and “season”视为固定效应。
--rand 6将“loc”视为随机效应。
对于多个性状,如果有一些环境因素需要作为模型效应进行拟合,那么所有性状都应该在模型效应中指定,并且请在一个性状中使用逗号作为分隔符,在性状之间使用空格,如果没有考虑到这些因素,则在一个性状中使用0,以固定效应为例:
./hiblup ... --dcovar 2,3 0 2,3,6 ...
第一个性状具有两个固定效应: “性别”和“季节”; 第二个性状没有固定效应; 第三个性状具有三个固定效应: “性别”、“季节”和“地点”。
重点
--qcovar 用于指定环境影响作为定量协变量,影响的记录应为数字,任何字符都不允许。
--dcovar用于将环境效应指定为离散的协变量,效应的记录可以是数字或字符,两者都是可接受的。
--rand指定环境随机效应,这些随机效应来自于记录的环境因素,遗传随机效应来自于家系或基因组信息,如果提供家系或基因型文件,HIBLUP 将自动适应模型中的遗传随机效应,用户不需要在这里添加个人 ID 列作为随机效应。
系谱文件
利用谱系文件导出分子关系矩阵和近交系数。文件中需要按顺序排列三列。第一列是个人 id,第二列和第三列分别是其父亲和母亲 id
NA Na . - NaN NAN nan na N/A n/a <NA>
与表型文件一样,上述列表中的任何符号出现在谱系文件中都将被视为缺失。家谱的示例文件如下:
id father mother
Ind2 NA NA
Ind5 Ind1 NA
Ind11 NA Ind2
Ind17 Ind11 Ind5
Ind22 Ind1 Ind5
Ind45 Ind22 Ind2
此文件需要使用参数项--pedigree。
注意
第一栏中的个人 ID 的顺序可以按出生日期排序,但这不是强制性要求,因为 HIBLUP 具有对输入谱系进行排序和重新排列的自动功能。
基因型文件
HIBLUP 目前只接受 PLINK 二进制格式的基因型,例如 demo.fam、 demo.bim 和 demo.bed,详情请参阅 PLINK 用户手册。用户可以通过 PLINK2和 TASSEL 将任何其他基因型格式(例如 VCF、 HapMap、 PED/MAP)转换为二进制格式:
# HapMap to VCF by TASSEL
./run_pipeline.pl -SortGenotypeFilePlugin -inputFile demo.hmp.txt
-outputFile demo.sort.hmp.txt -fileType Hapmap
./run_pipeline.pl -fork1 -h demo.sort.hmp.txt -export-exportType VCF -runfork1
# VCF to Binary
./plink2 --vcf demo.vcf --make-bed --out demo
# PED/MAP to Binary
./plink2 --ped demo.ped --map demo.map --make-bed --out demo
这些二进制文件需要分配参数项--bfile [prefix] (i.e., --bfile demo )
注意
(1) HIBLUP 只支持加载一个二进制文件,不支持加载多个二进制文件(例如,全基因组按染色体分别存储) ,请在使用 HIBLUP 之前通过 PLINK 进行合并,这里有一个粗略的指导。(https://www.biostars.org/p/148657/)
(2) HIBLUP 将 A1A1基因型编码为2,A1A2编码为1,A2A2编码为0,其中 A1是 *.bim文件中每个标记的第一个等位基因。因此,估计的效应大小是在 A1等位基因上,当一个过程涉及到标记效应时,用户应该注意它。
重点
HIBLUP 没有插补功能,二进制文件中的任何缺失基因型都将被视为杂合子,这意味着在分析中缺失将被迫编码为1,如果基因型中存在缺失,我们建议用户在使用 HIBLUP 之前通过其他软件进行插补。(例如beagle)
群体分层文件
由于不同的种群或品种有不同的遗传背景,因此不同种群的等位基因频率或基因型频率可能会有显著差异。HIBLUP 可以将种群分类信息整合到多种基因组分析中,例如等位基因或基因型频率计算、基因组关系矩阵构建、单个或多个性状模型拟合。人口分类文件编制简单,需要两栏,第一栏是个体ID列表,第二栏是个体所属的品种(以猪为例)。例如
ID Breed
Ind1 DD(杜洛克)
Ind2 YY(大白)
Ind3 LL(长白)
Ind4 YY
Ind5 LL
此文件需要指定参数项--pop-class
关系矩阵文件
HIBLUP 目前只接受二进制格式的关系矩阵(Relations Matrix,XRM) ,它具有最高的文件写入和数据加载效率,同时耗费最少的磁盘空间,例如 e.g. demo.GA.id
and demo.GA.bin。
文件 demo.GA.id 有一列列出所有个体的 id,即文件 demo.GA.bin 存储 XRM 的较低的三角形元素作为密集矩阵,以及文件demo.GA.bin 为稀疏矩阵存储 XRM 的行索引、列索引和三角形元素。
默认情况下,这些文件是由 HIBLUP 中的--make-xrm 生成的(参见构造关系矩阵)。
此外,我们还开发了一个文件格式转换器来制作这种二进制关系矩阵,请参阅相应的章节(见 XRM 的格式转换)。要使用 HIBLUP 的二进制 XRM,只需将其前缀分配给参数选项--xrm [prefix] :
./hiblup ... --xrm demo.GA ...
如果有多个 XRM,请使用逗号作为前缀之间的分隔符:
./hiblup ... --xrm demo.GA,demo.GD ...
重点
HIBLUP 具有计算 XRM 逆的功能,这可能对其他软件(如 BLUPF90,DMU,ASReml,…)有用,但对 HIBLUP 是无用的,因为 HIBLUP 使用 XRM 直接估计方差分量或解混合模型方程,因此请不要将关系矩阵的逆赋给 HIBLUP,除非你知道自己在做什么。
基因组估计育种值文件
基因组估计育种值(GEBVs)用于计算加性或显性 SNP 效应,第一列为样本 id,其余列为加性或显性 GEBVs,一个有三个个体的例子如下:
id add
Ind1 -1.1345
Ind2 0.3245
Ind3 0.0234
此文件需使用参数项--gebv
注意
如果指定了--add,则应该提供附加育种值,标志--dom 也应该提供附加育种值。如果同时指定了加法和加法,则应在文件中提供加性和显性的育种值,加性育种值的列应在显性育种值的前面。
SNP 加权文件
SNPs 加权文件用于构造加权 XRM 或计算 SNP 效应,第一列为 SNP id,其余列为 SNPs 的权值,请确保所有权值均为正值,具有三个 SNPs 的示例如下:
SNPid weight
SNP1 42
SNP2 0.3
SNP3 100
此文件需用参数项--snp-weight
注意
如果指定了—— add,则应提供附加权重,标志—— dom 也应提供附加权重。如果同时指定了 -add 和 -dom,则应在文件中提供所有加性和显性 SNP 的权重,加性权重列应位于显性权重的前面。
SNP 效应文件
SNP 效应文件用于预测基因型个体的 GEBV 或实施基因组交配,至少需要5列,举例如下:
SNPid a1 a2 freq_a1 add dom
SNP1 A C 0.1243 0.30134 0.00000
SNP2 G T 0.0345 -0.06324 0.00124
SNP3 A G 0.3635 0.15425 -0.00913
此文件应使用参数项--score
注意
如前所述,提供的单核苷酸多态性效应类型应该与其 -add 和 -dom一致,如果两者都提供,则加性单核苷酸多态性效应列应位于显性单核苷酸多态性效应的前列。
摘要数据文件
来自 gWAS/meta 分析的摘要数据应以 COJO 格式编制,见下文:
SNP A1 A2 FREQ BETA SE P NMISS
M1 G T 0.5181 -1.565 1.155 0.1762 500
M2 A G 0.145 -1.77 1.519 0.2445 500
M3 G A 0.3206 1.498 1.583 0.3445 500
M4 C G 0.5356 0.3366 1.003 0.7374 500
M5 C G 0.0975 1.27 1.755 0.4695 500
列为 SNP,效应等位基因,其他等位基因,效应等位基因的频率,效应大小,标准误差,p 值和样本量。注意“ A1”需要是效应等位基因,“ A2”是另一个等位基因,“ FREQ”应该是“ A1”的频率。
此文件需用参数项--sumstat
基因组窗口文件
HIBLUP 支持用户定义的窗口来切割基因组进行多种分析,例如 LD 和 LD 评分计算,SBLUP 模型拟合。需要三栏内容如下:
chr start stop
1 10583 1577084
1 1577084 2364990
1 2364990 3150345
1 3150345 4284187
1 4284187 4854314
2 10133 341834
2 341834 1161563
2 1161563 1688845
2 1688845 2829810
2 2829810 3389305
注意,窗口应该是非重叠的。
此文件需要参数项--window-file
样本和 SNP 过滤器文件
HIBLUP 可以过滤所有可用函数中的样本和 SNP,文件编写简单,只需在文件中列出个体名称或 SNP 的 ID,不需要头文件。
示例文件 id.txt
Ind16
Ind56
Ind110
如果用户需要在分析中删除这些个体,只需要将其指定为标记--remove。相反,如果它被分配到标记--keep,只有那些个人在文件中将用于分析。
示例文件snp.txt
SNP23
SNP198
SNP432
如果用户需要在分析中删除这些 SNP,只需要将其指定为标记--exclude。相反,如果将其分配给标记--extract,那么将只使用文件中的 SNP 进行分析。















 2406
2406











 被折叠的 条评论
为什么被折叠?
被折叠的 条评论
为什么被折叠?
 到【灌水乐园】发言
到【灌水乐园】发言









