







3.1多线程计算
我们已经实现了 HIBLUP 中的大多数过程能够在多个线程上运行。用户可以通过以下方法轻松地更改用于分析的线程数:
./hiblup --threads 32
如果没有指定-threads,HIBLUP 将尝试从标准的 OpenMP 环境变量中获取最大的线程数,并使用所有可用的资源来提高计算效率。
3.2等位基因频率和基因型频率
HIBLUP 可以计算标记的等位基因频率和基因型频率。
等位基因频率
如果在输入命令中检测到标志--allele-freq,HIBLUP 将为基因型中的所有 SNP 计算 a 1等位基因频率:
./hiblup --bfile demo --allele-freq --out demo
它将生成一个名为“ demo.afreq”的文件,该文件的简要视图如下:
SNP a1 a2 freq_a1
snp1 A C 0.2775
snp2 G T 0.4475
snp3 A G 0.1918
然后用户可以过滤具有特定条件的 SNP,例如,在0.05 < freq < = 0.1的条件下拾取 SNP:
awk '$4 > 0.05 && $4 <= 0.1 {print $1}' demo.afreq > freq_filter_snp.txt
HIBLUP 还可以支持计算子群的等位基因频率,假设二进制基因型文件中有混合群,我们对群体之间的分布或比较等位基因频率的差异感兴趣,在运行 HIBLUP 时,通过指定一个群体分类文件(见文件格式)到选项“--pop-class很容易实现,例如:
./hiblup --bfile demo --allele-freq --pop-class demo.popclass.txt --out demo
然后,不同组别的等位基因频率将按栏目列出:
SNP a1 a2 DD_freq_a1 YY_freq_a1 LL_freq_a1
snp1 A C 0.2353 0.1245 0.5423
snp2 G T 0.1235 0.3456 0.2356
snp3 A G 0.0235 0.1224 0.2123
基因型频率
如果在输入命令中检测到标志--geno-freq,HIBLUP 将计算基因型中所有单核苷酸多态性的 A1A1和 A2A2基因型频率:
./hiblup --bfile demo --geno-freq --out demo
它将生成一个名为 demo.gfreq 的文件,该文件的简要视图如下:
SNP a1 a2 freq_a1a1 freq_a2a2
snp1 A C 0.3456 0.1244
snp2 G T 0.2358 0.2345
snp3 A G 0.1395 0.4643
如上所述,HIBLUP 还支持计算子组的基因型频率,只需在运行--geno-freq时指定一个人口分类文件到选项-pop-class。
3.3纯合与杂合
纯合性和杂合性被定义为个体的纯合性和杂合性的比例,它可以很容易地从 HIBLUP 中分别使用标志--homo 和--hete 获得。
纯合性
./hiblup --bfile demo --homo --out demo
一个名为 demo.homo 的文件将生成如下:
id a1a1 a2a2
IND0701 0.42255 0.201235
IND0702 0.415291 0.204923
IND0703 0.426455 0.199282
杂合性
./hiblup --bfile demo --hete --out demo
将生成一个名为 demo.hete 的文件,如下所示:
id a1a2
IND0701 0.376216
IND0702 0.379786
IND0703 0.374263
3.4构造关系矩阵(XRM)(重点重点重点)
HIBLUP 可以根据家系和基因组信息构建 XRM,输出的二进制文件有两种存储格式。对于稠密矩阵,关系矩阵下三角形的值按照“ ijk”格式在二进制文件中存储,一个元素占用超过4个字节,而对于稀疏矩阵,只有关系矩阵下三角形的非零值(k)及其行(i)和列索引(j)按照“ ijk”格式存储,即一个元素占用超过12个字节。
基于谱系的关系矩阵(PRM)
# construct A and its inverse simultaneously
./hiblup --make-xrm
--pedigree demo.ped
--add
--add-inv #wether to construct its inverse matrix
--thread 32
--out demo
名为 demo.PA.bin、 demo.PA.id、 demo.PAinv.sparse.bin、 emo.PAinv.sparse.id 的文件将在 work 目录中生成。
由于二进制文件(*.bin)不能直接打开,用户可以在命令中添加一个标志--write-txt,以输出一个可读的.txt文件。
有时,在计算 XRM 的反向(逆?)时,可能会遇到一个错误:
Error: matrix is not invertible,…
这是因为矩阵不是正定(positive-defined)的,用户可以通过岭回归(ridge regression method)的方法指定一个小的值--ridge-value来解决这个问题。
Genome based relationship matrix (GRM)
关系矩阵也可以从全基因组标记得到。构建基因组关系矩阵(GRM)的数学公式如下:
 其中
其中 Z 是数字编码的基因型矩阵,其维数为 n*m,n 是个体数,m是标记数,tr 是矩阵的轨迹。在 HIBLUP 中,总共有4种方法可以获得Z的加性和显性遗传效应,不同方法的基因型编码方法可参见下表:
 其中
其中 pA 和 pa 分别是 A 和 a 的等位基因频率。pAA、 pAa 和 paa 分别是 AA、 Aa 和 aa 的基因型频率。
用户可以通过指定具有相应值的标志--code-method,例如--code-method 2,轻松切换到上述任何一种方法。不同方法的参考资料见下列文件:
--code-method 1, (default) Su, Guosheng, et al. “Estimating additive and non-additive genetic variances and predicting genetic merits using genome-wide dense single nucleotide polymorphism markers.” Plos one (2012): e45293.
--code-method 2, Zeng, Zhao-Bang, Tao Wang, and Wei Zou. “Modeling quantitative trait loci and interpretation of models.” Genetics 169.3 (2005): 1711-1725.
--code-method 3, Yang, Jian, et al. “Common SNPs explain a large proportion of the heritability for human height.” Nature genetics 42.7 (2010): 565-569.
--code-method 4, Vitezica, Zulma G., et al. “Orthogonal estimates of variances for additive, dominance, and epistatic effects in populations.” Genetics 206.3 (2017): 1297-1307.
普通 GRM
构建加性和显性基因组关系矩阵的脚本:
# construct A and D simultaneously
./hiblup --make-xrm --code-method 2
--bfile demo
--add --dom
--step 10000
--thread 32
--out demo
#--write-txt #to output a readable ".txt" file
名为 demo.GA.bin、 demo.GA.id、 demo.GD.bin、 demo.GD.id 的文件将在 work 目录中生成,HIBLUP 将直接计算 GRM 的逆,并在命令中检测到--add-inv 或--dom-inv 时输出它。
注意,用户可以将列出 SNP 名称的文件分配给标志--extract或--exclude,以便通过 SNP 的子集构造 GRM。例如,构建 GRM 只使用重要的 SNP:
./hiblup --make-xrm
--bfile demo
--add
--extract sig.snp.txt
--thread 32
--out demo
其中,sig.snp.txt 文件包含被认为与某个特征显著相关的 SNP。
多种群的混合 GRM
如果基因型数据有多个群体(例如不同品种) ,HIBLUP 可以构建混合 GRM,混合 GRM 的数学公式可以表示如下:
 where ZiZi and ZjZj are the centered or scaled genotype matrix (as shown in above Table 1) using corresponding allele frequency or genotype frequency of ithith and jthjth population, respectively. nini and njnj are the sample size of different populations.(翻不好哈哈哈哈将就着看,)
where ZiZi and ZjZj are the centered or scaled genotype matrix (as shown in above Table 1) using corresponding allele frequency or genotype frequency of ithith and jthjth population, respectively. nini and njnj are the sample size of different populations.(翻不好哈哈哈哈将就着看,)
在运行 HIBLUP 构造常规 GRM 时,只需提供一个种群分类文件(见文件格式) ,就可以很容易地实现:
# construct mixed GRM
./hiblup --make-xrm --code-method 1
--bfile demo
--pop-class demo.popclass.txt
--step 10000
--thread 32
--out demo
加权 GRM
HIBLUP 支持通过以下脚本计算加权 GRM:
./hiblup --make-xrm --code-method 1
--bfile demo
--add --dom
--snp-weight snp.wt.txt
--thread 32
--out demo
SNP 权重的文件格式请参考这里(http://hiblup.com/tutorials#snps-weighting-file)。
基于谱系和基因组的关系矩阵(HRM)
当同时提供家系和基因组信息时,HIBLUP 将根据下面的方程自动构建 H 矩阵(Aguilar 2010,Christensen 2010) :
 其中
其中 A11和 A22分别是非基因型个体和基因型个体 A 矩阵的子矩阵,A12和 A21是 AA 矩阵的子矩阵,代表基因型个体和非基因型个体之间的关系,Gw = (1-alpha) * Gadj + alpha * A22Gw = (1-alpha) * Gadj + alpha * A22,Gadj 是从原始基因组关系矩阵 G 调整的简单线性回归:
那么我们有 Gadj = G * a + b。
./hiblup --make-xrm
--pedigree demo.ped
--bfile demo
--alpha 0.05
--add --dom
--out demo
上面的命令将同时生成加性关系矩阵和主导关系矩阵,文件名为: demo.HA.bin、 demo.HA.id、 demo.HD.bin、 demo.HD.id。
用户还可以指定--snp-weight 来构造加权 GRM 或指定--pop-class 来构造混合 GRM 来构造 HRM。
除了提供二进制基因型来构建 HRM 外,用户还可以使用自己预先计算的 GRM 来构建 HRM,但预先计算的 GRM 应该以二进制格式存储,可以通过 HIBLUP (参见 XRM 的格式转换)从正方形或三角形文本文件转换,相应的命令如下:
./hiblup --make-xrm
--pedigree demo.ped
--xrm yourGRM # yourGRM.bin, yourGRM.id
--alpha 0.05
--add # or '--dom' is supported
--out demo
此外,HIBLUP 还可以通过下面的脚本构造 H 矩阵的逆:
./hiblup --make-xrm
--pedigree demo.ped
--bfile demo
--alpha 0.05
--add-inv --dom-inv
--out demo
生成的文件名是: demo.hainv.Sparse.bin, demo.hainv.sparse.id ,demo.HDinv.parse.bin, demo.hd.sparse.id。
如上所述,您还可以使用自己的预计算 GRM 构造 HRM 的逆。
环境随机效应的 XRM
除了为遗传随机效应构建 XRM 外,HIBLUP 还可以为环境随机效应构建 XRM,为此应提供表型文件,命令如下:
./hiblup --make-xrm
--pheno demo.phe
--rand 6,7
--out demo
他生成的文件名是: demo.loc.Sparse.bin, demo.lco.sparse.id ,demo.dam.parse.bin, demo.dam.sparse.id。
注意
(1)上述关系矩阵,除了前缀含有“.sparse.”的关系矩阵除外。可用于 GCTA,LDAK,但需要额外的过程,例如:
awk '{print $1,$1}' demo.GA.id > gcta.grm.id
cp demo.GA.bin gcta.grm.bin
# then use it by GCTA
./gcta64 ... --grm gcta ...
(2)由 GCTA 或 LDAK 计算的二进制格式关系矩阵可以直接由 HIBLUP 使用,无需任何格式转换,只需将其前缀指定为标志--xrm 即可。
3.5. XRM 的格式转换
HIBLUP 只接受二进制文件格式的 XRM,因为一些软件(如 DMU,BLUPF90,ASReml)不能识别它,而且,编程初学者很难准备这些二进制文件,因此我们提供了一个格式转换器,可以方便地将 HIBLUP 与其他软件连接起来。
二进制转换为文本
下面的命令将 XRM 以文本格式输出为一个正方形矩阵,名称为 demo.txt 和 demo.id.txt:
./hiblup --trans-xrm
--xrm demo.GA
--out demo
# --triangle
一个包含三个人的 demo.txt 示例如下:
1.55755 1.04847 0.64622
1.04847 1.58144 0.66971
0.64622 0.66971 1.56794
如果检测到标志--triangle,HIBLUP 将只将矩阵的下三角形写入格式为 < i j k > 的文本文件中,其中 i、 j、 k 分别为行、列索引及其对应值。对于稀疏矩阵,HIBLUP 在输出“ ijk”格式时会忽略0个元素。上述三个人的示例文件如下:
文本转换为二进制
如果用户有自己的文本格式的先验计算关系矩阵,HIBLUP 可以将其转换成二进制文件,HIBLUP 可以直接使用,该脚本如下:
./hiblup --trans-xrm
--xrm-txt demo.txt
--xrm-id demo.id.txt
--out demo
以上命令需要正方形的 demo.txt 和 demo.id.txt。如果文件 demo.txt 是“ ijk”格式,请记住添加标志--triangle。
注意
HIBLUP 还可以将 GCTA、 LDAK 计算的关系矩阵转换为文本格式:
./hiblup --trans-xrm --xrm gcta.grm --out hiblup
3.6计算近亲繁殖和亲缘关系系数
利用家系或基因型信息导出近交系数和亲缘关系系数,HIBLUP 首先构造加性分子矩阵,然后在文件中导出并输出系数。近交系数和亲缘系数的标志分别是--ibc 和--rc。
近亲繁殖系数
# derived from pedigree
./hiblup --ibc --pedigree demo.ped --thread 32 --out demo
# or derived from prior calculated PA matrix
./hiblup --ibc --xrm demo.PA --out demo
# derived from genotype
./hiblup --ibc --bfile demo --thread 32 --out demo
# or derived from prior calculated GA matrix
./hiblup --ibc --xrm demo.GA --out demo
近亲繁殖系数保存在 demo.ibc 文件中:
id ibc
IND0702 0.00290048
IND0703 0.00262272
IND0704 0.00262272
亲缘关系系数
# derived from pedigree
./hiblup --rc --pedigree demo.ped --thread 32 --out demo
# or derived from prior calculated PA matrix
./hiblup --rc --xrm demo.PA --out demo
# derived from genotype
./hiblup --rc --bfile demo --thread 32 --out demo
# or derived from prior calculated GA matrix
./hiblup --rc --xrm demo.GA --out demo
关系系数保存在 demo.rc 文件中:
id1 id2 rc
IND0701 IND0701 1
IND0701 IND0702 0.472652
IND0701 IND0703 0.0721178
IND0701 IND0704 0.0721178
注意
- 近亲繁殖系数和亲缘关系系数可以同时计算,如:
./hiblup --ibc --rc --pedigree demo.ped --out demo
./hiblup --ibc --rc --bfile demo --out demo
2.基因组近交系数和亲缘关系系数可以用不同的方法计算,使用旗码法,更详细的可以在这里看到(https://www.hiblup.com/tutorials#construct-relationship-matrix-xrm)。
./hiblup --ibc --rc --bfile demo --code-method 1 --out demo
3.7主成分分析
HIBLUP 具有计算种群主成分分析的功能,应提供基因型或关系矩阵。
从基因型计算 PCs
./hiblup --pca --bfile demo --out demo
注意,用户可以添加标志--step n 来控制内存成本,并指定标志--npc n 来输出几个而不是所有的 PCs。
从 XRM 计算 PC
./hiblup --pca --xrm demo.GA --npc 5 --out demo
将生成两个文件 demo.pc 和 demo.pcp。
文件 demo.pc 包含计算出的 PC,文件概述:
id PC1 PC2 PC3 PC4 PC5
IND0701 0.0413326 -0.00664808 -0.00795809 0.0218009 -0.0156735
IND0702 0.00856316 0.0378125 0.000503819 0.0205049 0.00972465
IND0703 -0.00793242 0.0458023 -0.00438572 -0.0173733 -0.0254701
IND0704 -0.00793248 0.0458023 -0.00438575 -0.0173733 -0.0254701
文件 demo.pcp 包含 PC 的比例和累积比例,文件概述:
Components PC1 PC2 PC3 PC4 PC5
Standard deviation 4.23976 4.10211 3.76024 3.66821 3.60725
Proportion of Variance 0.0224695 0.0210342 0.0176743 0.0168197 0.0162653
Cumulative Proportion 0.0224695 0.0435037 0.0611779 0.0779976 0.094263
3.8. LM、 BLUP、 PBLUP、 GBLUP (rrBLUP)、 SSGBLUP
用户可以方便地实现简单线性模型(LM)、 BLUP、 PBLUP、 GBLUP (rrBLUP)和 SSGBLUP,HIBLUP 可以根据用户提供的数据类型自动切换到其中一种。下表详细说明了 HIBLUP 中不同型号所需或不需要的标志:
 RrBLUP 模型在理论上与 GBLUP 模型相同,用户只需在 GBLUP 模型的基础上添加一个标志
RrBLUP 模型在理论上与 GBLUP 模型相同,用户只需在 GBLUP 模型的基础上添加一个标志--snp-effect,即可获得 rrBLUP 模型的 SNP 效果。
重点
值得注意的是,由于 FSPAK 技术在非常稀疏的混合模型方程“ LHS”上的高性能,PBLUP 模型可以非常有效。然而,HIBLUP 是为基于基因组的遗传评估而设计的,其中“ LHS”不再稀疏,在 HIBLUP 中实现的基于“ V”矩阵的策略将更适合这种情况。虽然用户可以通过使用 HIBLUP 来适应 PBLUP 模型,但是与 DMU、 BLUPF90等软件相比,它可能会更慢,并且耗费更多的内存。因此,我们建议使用 DMU 或 BLUPF90,当拟合模型只涉及血统信息。
3.9单性状模型
单性状模型旨在估计一个性状的方差分量及其所有随机效应的遗传性。永远记住,没有必要调整个人或标记的顺序,以保持不同文件之间的一致性,HIBLUP 可以自动做到这一点。对单一特征模型运行 SSGLBUP 的命令示例:
./hiblup --single-trait
--pheno demo.phe
--pheno-pos 8
--dcovar 2,3 #fixed effect
--qcovar 4,5 #covariates
--rand 6,7 #non-genetic (environmental) random effect
--pedigree demo.ped #genetic random effect
--bfile demo #genetic random effect
--add --dom
--threads 32
--out demo
输入文件的详细格式请参考表型、基因型。如何设置多个性状的协变量(--qcovar)、固定(--dcovar)和随机(--rand)效应,请参考这里https://http//www.hiblup.com/tutorials#covariates-fixed-and-environmental-random-effects。
注意,支持 XRM 构造的所有参数(例如,加权 GRM、混合 GRM 或 HRM,…)也可以应用于上面的命令。
方差分量估计有5种算法:
“AI”、“ EM”、“ HE”、“ EMAI”、“ HI”,其中“ AI”为 AIREML,“ EM”为 EMREML,“ HE”代表 HE 回归,“ HI”为“ HE + AI”,使用 HE 回归的结果作为 AIREML 的起始值,用户可以通过标志---vc-method选择其中之一,并通过标志--ai-maxit 和--em-maxit 分别更改 AIREML 和 EMREML 的最大迭代次数。对于“ HE”回归,如果提供固定效应的协变量,HIBLUP 将首先回归这些表型,然后使用残差进行 HE 回归,HE 的标准误差(SE)通过 Jack析重复100次的重采样策略计算。默认情况下,HIBLUP 不计算 HE 回归的随机效应,用户可以添加一个标志--he-pred 来输出它,只有在这种情况下,标志--pcg 才能用于非常大的数据集上的快速计算,我们称之为“ HE + PCG”策略。
然而,上面的命令在逐个特征拟合特征时可能会很费时间,因为它会为每个特征重复构造 XRM。我们只能在开始时构造一次 XRM,然后在适应多个 trait 时将其赋值给--xrm,这样可以得到相同的结果,但是效率会高得多,命令可以如下所示:
# Step 1: construct XRMs
./hiblup --make-xrm
--pedigree demo.ped
--bfile demo
--add --dom
--out demo
# Step 2: link XRMs to fit model
./hiblup --single-trait
--pheno demo.phe
--pheno-pos 8
--dcovar 2,3
--qcovar 4,5
--rand 6,7
--xrm demo.HA,demo.HD
--add --dom
--threads 32
--out demo
将在工作目录中生成几个文件:
demo.vars: 所有随机效应的估计方差和遗传性:
Var Var_SE h2 h2_SE
loc 12.2127 8.9109 0.1035 0.0682
dam 10.6268 4.8605 0.0901 0.041
HA 59.2007 11.7641 0.5019 0.0807
HD 28.9946 8.9294 0.2458 0.083
e 6.9232 1.6957 0.0587 0.0161
demo.beta: 所有协变量和固定效应的估计系数:
Levels Estimation SE
mu 32.5285 3.4758
sex_F 6.2933 1.7593
sex_M 26.2353 1.7634
season_Autumn 18.0905 0.9753
season_Spring -1.9168 0.9904
season_Summer 7.8471 1.0066
season_Winter 8.5078 0.9944
day 0.1547 0.0574
bornweight 1.5703 0.4614
demo.anova: 所有协变量和固定效应的方差分析表:
Factors Df SumSq MeanSq F Pr(>F)
sex 1 49710.3845 49710.3845 7180.311 <2e-16
season 3 24515.0941 8171.698 1180.344 <2e-16
day 1 925.3352 925.3352 133.658 <2e-16
bornweight 1 437.3432 437.3432 63.171 1.30E-14
e 493 3413.1141 6.9232
demo.rand: 估计的环境随机效应,遗传随机效应(名为“ PA”,“ GA”或“ HA”的列是表型和非表型个体的育种值的列表)和残差。
用户可以指定所有未知方差的起始值到--vc-priors,以便快速收敛,如何输入方差分量的方法可以在这里找到http://hiblup.com/tutorials#solve-mixed-model。
3.10. 单性状模型的重复记录
运行重复记录模型与拟合单性状模型几乎相同,唯一的区别是重复记录模型能够拟合永久环境效应,这可以通过在表型文件的第一列分配个体 id 来实现环境随机效应,以 GBLUP 模型为例:
./hiblup --single-trait
--pheno demo.repeat.phe
--pheno-pos 2
--rand 1
--bfile demo
--threads 32
--out demo
应该指出的是,HIBLUP 在遗传参数估计中使用表型记录的方差-协方差矩阵,因此如果存在低重复(例如来自1000个个体的2000个记录重复两次) ,HIBLUP 将比基于 MME 的软件更快。但是如果被分析的特征被高度复制(例如,来自200个个体的2000条记录被复制10次) ,BLUPF90,ASreml 或其他基于 MME 的算法将会更快,我们建议你使用这个软件。
3.11多性状模型
多性状模型用于估计性状间的遗传相关性,HIBLUP 分析中使用的性状数目不受限制。此外,用户可以指定不同的协变量,固定和随机效应的不同性状。应该指出的是,HIBLUP 现在只能处理平衡的数据,这意味着只有个人有效记录的所有特征将用于分析。PBLUP、 GBLUP、 SSGBLUP 都可用于多性状模型,HIBLUP 可根据提供的文件自动切换到其中一个。永远记住,没有必要调整个人或标记的顺序,以保持不同文件之间的一致性,HIBLUP 可以自动完成。
以 GBLUP 为例,多模型可以通过以下命令实现:
./hiblup --multi-trait
--pheno demo.phe
--pheno-pos 8 9 10
--qcovar 4,5 5 4
--dcovar 2,3 0 2
--rand 6,7 7 0
--xrm demo.GA,demo.GD # same with --bfile demo --add --dom
--vc-method AI
--ai-maxit 30
--threads 32
--out demo
输入文件的详细格式请参考表型、基因型。如何设置多个性状的协变量(--qcovar)、固定(--dcovar)和随机(–rand)效应,请参考这里http://hiblup.com/tutorials#covariates-fixed-and-environmental-random-effects。
方差分量估计有5种算法:
“AI”、“ EM”、“ HE”、“ EMAI”、“ HI”,其中“ AI”为 AIREML,“ EM”为 EMREML,“ HE”代表 HE 回归,“ HI”为“ HE + AI”,使用 HE 回归的结果作为 AIREML 的起始值,用户可以通过标志---vc-method选择其中之一,并通过标志--ai-maxit 和--em-maxit 分别更改 AIREML 和 EMREML 的最大迭代次数。对于“ HE”回归,如果提供固定效应的协变量,HIBLUP 将首先回归这些表型,然后使用残差进行 HE 回归,HE 的标准误差(SE)通过 Jack析重复100次的重采样策略计算。默认情况下,HIBLUP 不计算 HE 回归的随机效应,用户可以添加一个标志--he-pred 来输出它,只有在这种情况下,标志--pcg 才能用于非常大的数据集上的快速计算,我们称之为“ HE + PCG”策略。
将生成几个文件:
demo.vars:所有性状的所有随机效应的方差、遗传力和 SE:
Item Var Var_SE h2 h2_SE
tr1_loc 13.73854 9.9309 0.11739 0.07538
tr1_dam 2.29288 3.18932 0.01959 0.02731
tr1_demo.GA 61.35173 9.57171 0.52423 0.06964
tr1_demo.GD 29.93077 6.27996 0.25575 0.06022
tr1_e 9.71917 1.67088 0.08305 0.01696
tr2_dam 3.42172 3.80424 0.02971 0.03308
tr2_demo.GA 63.59 10.52058 0.55219 0.06017
tr2_demo.GD 38.14332 7.27491 0.33122 0.06696
tr2_e 10.00395 1.63578 0.08687 0.01578
tr3_demo.GA 21.03906 6.94184 0.2203 0.0668
tr3_demo.GD 15.25593 10.56861 0.15974 0.10919
tr3_e 59.20689 9.04084 0.61996 0.09801
demo.covars: 所有遗传随机效应的所有性状的协方差、相关性和 SE:
Item COVar COVar_SE r r_SE
tr1:tr2_demo.GA 46.6899 8.56852 0.747524 0.171364
tr1:tr3_demo.GA 16.3376 5.96333 0.454749 0.185599
tr2:tr3_demo.GA 12.3489 6.21401 0.337612 0.157671
tr1:tr2_demo.GD 26.9273 4.65452 0.796922 0
tr1:tr3_demo.GD -2.25039 4.95673 -0.105301 0.23413
tr2:tr3_demo.GD 8.23124 5.21494 0.341194 0.259133
tr1:tr2_e 0.24039 1.17428 0.024379 0.119333
tr1:tr3_e -1.10713 2.87487 -0.0461537 0.11985
tr2:tr3_e -4.80127 2.85341 -0.197284 0.128375
demo..anova: 协变量的方差分析表,不同性状的固定效应。
demo..beta: 协变量的估计系数和 SE,不同性状的固定效应。
demo.*.rand: 估计的环境随机效应,遗传随机效应(列名为“ PA”,“ GA”或“ HA”是表型和非表型个体的育种值列表)和残差。不同的特质。
用户可以将所有未知方差和协方差的起始值指定为--vc-priors和--covc-priors,如何输入遗传成分的方法可以在这里找到。
注意
HIBLUP 仅估计遗传随机效应和残差效应的协方差,而不是环境随机效应,因此模型中至少应该包括一个遗传随机效应,可以来自谱系(--pedigree) ,基因型(--bfile)或 XRM (--xrm)。如果用户坚持估计环境随机效应的相关性,只需要通过标记为这些随机效应建立关系矩阵--make-xrm (请参阅“环境随机效应的 XRM”一章) ,然后将其分配给选项--xrm。
3.12. 求解混合模型方程
求解混合模型的目的是利用已知的遗传参数估计所有个体的育种值。HIBLUP 根据指定的标志--pheno-pos 的长度自动切换到单个性状或多个性状模型。永远记住,没有必要调整个人或标记的顺序,以保持不同文件之间的一致性,HIBLUP 可以自动做到这一点。
以 GBLUP 模型为例,对单性状和多性状模型的命令如下:
# (1) additive effect for single trait model
./hiblup --mme
--pheno demo.phe
--pheno-pos 8
--rand 6
--xrm demo.GA # can be replaced by --bfile demo --add
--vc-priors v1,v2,v3
--pcg
--threads 32
--out demo
# (2) additive and dominant effect for multiple traits model
./hiblup --mme
--pheno demo.phe
--pheno-pos 8 9 10
--rand 6,7 7 0
--bfile demo # can be replaced by --xrm demo.GA,demo.GD
--add --dom
--vc-priors t1_v1,t1_v2,t1_v3,t1_v4,t1_v5 t2_v1,
t2_v2,t2_v3,t2_v4 t3_v1,t3_v2,t3_v3
--covc-priors cov1,cov2,cov3 cov4,cov5,cov6 cov7,cov8,cov9
--pcg
--threads 32
--out demo
在缺省情况下,HIBLUP 直接计算 V 矩阵的逆,用户可以尝试添加标志--pcg,使用预条件共轭梯度算法求解线性方程组,在某些情况下更快。
重点
(1)--vc-priors包含所有随机效应的方差分量,包括环境随机效应、遗传随机效应和残差,所提供的方差分量也应该是这个顺序。
对于单个 trait 模型,请使用逗号作为分隔符,如第一个示例命令所示:
v1: the variance of the environmental random effect located
at the 6th column in phenotype file.
v2: the additive genetic variance.
v3: the residual variance.
对于多性状模型,方差分量按性状顺序排列,请在一个性状中使用逗号,在性状之间使用空格,如第二个例子命令所示:
t1_v1: the variance of the environmental random effect located
at the 6th column in phenotype file for the first trait.
t1_v2: the variance of the environmental random effect located
at the 7th column in phenotype file for the first trait.
t1_v3: the variance of additive genetic effect for the first trait.
t1_v4: the variance of dominant genetic effect for the first trait.
t1_v5: the variance of residuals for the first trait.
t2_v1: the variance of the environmental random effect located
at the 7th column in phenotype file for the second trait.
t2_v2: the variance of additive genetic effect for the second trait.
t2_v3: the variance of dominant genetic effect for the second trait.
t2_v4: the variance of residuals for the second trait.
t3_v1: the variance of additive genetic effect for the third trait.
t3_v2: the variance of dominant genetic effect for the third trait.
t3_v3: the variance of residuals for the third trait.
(2)--covc-priors包含所有遗传随机效应中性状之间的协方差成分,协方差成分按遗传随机效应顺序排列,请在遗传随机效应中使用逗号,在遗传随机效应中使用空格,输入顺序如下:
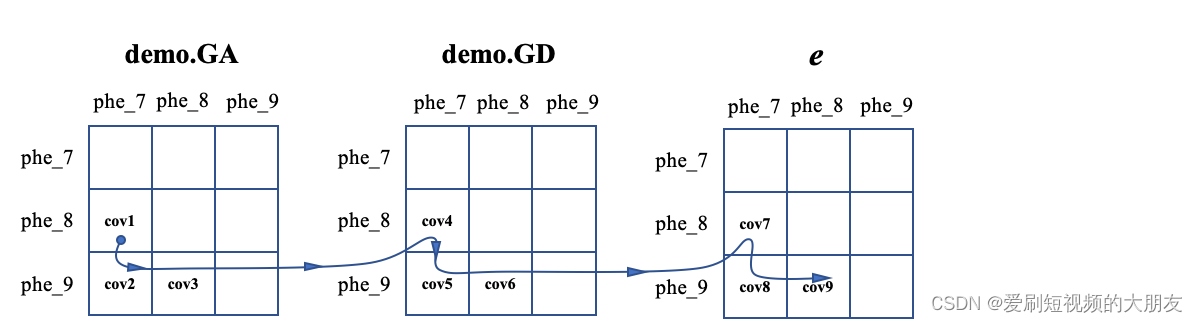 其中每个盒子代表一个遗传随机效应,非对角线是这个遗传随机效应中性状之间的协方差,而对角线是它在不同性状中的方差。Box 的数目等于 XRM 的数目加1,其中“1”是余数。用户需要在每个盒子的下三角形处给 HIBLUP 赋值:
其中每个盒子代表一个遗传随机效应,非对角线是这个遗传随机效应中性状之间的协方差,而对角线是它在不同性状中的方差。Box 的数目等于 XRM 的数目加1,其中“1”是余数。用户需要在每个盒子的下三角形处给 HIBLUP 赋值:
cov1: the co-variance between the first and second trait at "demo.GA".
cov2: the co-variance between the first and third trait at "demo.GA".
cov3: the co-variance between the second and third trait at "demo.GA".
...
输入协方差的顺序应严格遵循上图中的路线。
3.13拟合影响的相互作用
环境相互作用(E 由 E)
记录环境之间的相互作用可以作为固定或随机效果,请使用冒号作为分隔符。
如装配为固定效果:
./hiblup ... --dcovar 2,2:3:6
上述指令表明,这一特征的固定效应是“性别”,即“性别”、“季节”、“场所”之间的相互作用。
如果是随机效果:
./hiblup ... --rand 6,6:7,7
同样,这种性状的随机效应是“ loc”,即“ loc”与“ dam”、“ dam”之间的相互作用。
对于多性状模型,使用空格作为分隔符,以3个性状为例:
./hiblup ... --dcovar 2:3 0 2,3,2:3 --rand 6 7,6:7 0
遗传相互作用(G 由 G)
HIBLUP 可以拟合遗传随机效应的相互作用,例如: 加性与加性、加性与显性、显性与显性等,在一个相互作用项上遗传随机效应的数目是不受限制的。在 HIBLUP 中 G 与 G 对 n 个遗传随机效应的相互作用可以从数学上表述如下:

其中 N 是个体的个数,点圈圈 是所有矩阵的 Hadamard 乘积(即元素乘法) ,tr ()是矩阵的轨迹。
适合 G 的唯一方法是指定随机效果的前缀来标记--xrm,请在交互术语中使用冒号作为分隔符:
# fit A by D only
./hiblup ... --xrm demo.GA:demo.GD
#fit A, D, and A by D
./hiblup ... --xrm demo.GA,demo.GD,demo.GA:demo.GD
#fit A by A, A by D and D by D simultaneously
./hiblup ... --xrm demo.GA:demo.GA,demo.GA:demo.GD,demo.GD:demo.GD
遗传-环境相互作用(G 比 E)
遗传-环境相互作用将作为随机效应包含在模型中,请为环境因素在表型文件中指定一个代表其位置的数字值,并为标志“--rand-gxe”指定遗传效应的前缀。
对于单性状模型,请在一个交互术语中使用冒号作为分隔符,并在多个交互术语中使用逗号作为分隔符,例如:
./hiblup --single-trait ... --rand-gxe 6:demo.GA,6:demo.GD
#interaction among multiple effects
./hiblup --single-trait ... --rand-gxe 6:demo.GA:demo.GD
./hiblup --single-trait ... --rand-gxe 6:7:demo.GA
对于多性状模型,请使用性状之间的空间,如果一个性状没有 G 到 E 的交互作用,请使用0,以3个性状为例:
./hiblup --multi-traits ... --rand-gxe 6:demo.GA 6:demo.GA,7:demo.GA 0
以上命令表示第一性状的交互作用是加性遗传效应的“ loc”; 第二性状的交互作用是加性遗传效应的“ loc”和加性遗传效应的“ dam”; 第三性状不考虑交互作用。
3.14效应显著性的统计检验
对于固定效应和协变量,HIBLUP 可以自动进行统计检验,并将结果列入文件 *.anova 。最后一列 F 检验的 p 值可以作为一个诊断,其中固定效应或协变量对一个性状具有统计学意义。
对于随机效应(遗传或环境) ,没有显著性统计检验的自动功能,用户可以通过下列步骤手动进行似然比检验(LRT) :
(1)利用感兴趣的随机效应运行完整的模型,得到类日志值 L1(可以在 REML 的最后一次迭代中从 HIBLUP 的日志信息中找到)。
(2)运行减少利率随机效应的约简模型,得到类日志值 L0。
(3)在 R 中用卡方检验计算 p 值:
> stat <- 2 * (L1 - L0)
> pvaue <- pchisq(stat, df = 1, lower.tail = FALSE)
> print(pvaue)
3.15可靠性及PEV
对于单性状模型、多性状模型和求解混合模型方程,HIBLUP 可以计算所有随机效应的信度和预测误差方差(PEV)。可靠性计算的数学公式如下:

其中 σ2是利率随机效应的 REML 分析的估计方差。预测误差方差 PEV 可以从以下方程得到:

其中 σe2是估计的残差方差。Cii 是混合模型方程左侧逆矩阵(LHS)的第二对角元。
缺省情况下,HIBLUP 不计算和输出这些统计数据,因为可靠性估计通常比整个 REML 过程花费更长的时间,因此请添加标志--r2只有当您真正想要得到可靠性时才添加。
以单性状模型为例,给出了获得可靠性的步骤:
./hiblup --single-trait
--pheno demo.phe
--pheno-pos 8
--dcovar 2,3 #fixed effect
--qcovar 4,5 #covariates
--rand 7 #non-genetic (environmental) random effect
--bfile demo #genetic random effect
--r2 #to calculate the reliability
--threads 32
--out demo
然后,每个随机效应,包括残差,有几个额外的列列出其相应的标准错误,PEV 和可靠性,文件概述:
ID dam dam_SE dam_PEV dam_R2 GA GA_SE GA_PEV GA_R2 residuals residuals_SE residuals_PEV residuals_R2
IND1001 0.496645 3.75992 14.137 0.255863 6.66127 4.27597 18.2839 0.774767 -1.45194 3.18313 10.1323 0.55248
IND1002 0.496645 3.75992 14.137 0.255863 6.66127 4.27597 18.2839 0.774767 2.04383 3.17906 10.1064 0.553624
IND1003 0.674833 3.53678 12.5088 0.341566 2.08201 4.62091 21.3528 0.736963 -0.104053 4.14567 17.1866 0.24091
IND1004 0.674833 3.53678 12.5088 0.341566 -0.682836 4.70199 22.1087 0.727651 0.9083 4.17824 17.4577 0.228936
3.16计算 SNP 效应
HIBLUP 现在可以计算所有 SNP 的加性效应和显性效应。有两种方法可以计算 SNP 效应,第一种方法是在命令中添加标志--snp-effect,用于拟合单个性状、多个性状模型或求解混合模型:
# compute additive SNP effect by SSGBLUP of single trait model
./hiblup --single-trait
--pheno demo.phe
--pheno-pos 8
--bfile demo
--pedigree demo.ped
--add
--snp-effect
--thread 32
--out demo
第二种方法是从先前计算的 GEBV 推导出 SNP 效应:
# compute dominant SNP effect
./hiblup --snp-effect
--gebv demo.gebv.d.txt
--bfile demo
--dom
--thread 32
--out demo
GEBV 的文件格式可以在这里找到http://116.62.61.25/tutorials#genomic-estimated-breeding-value-file。由 --bfile 指定的基因型文件被用来构建 GRM 和计算 SNP 效应,第二种方式,HIBLUP 将为个体构建加性或显性 GRM,为了节省时间,用户可以提供事先计算的 GRM,HIBLUP 将直接使用它:
# compute additive and dominant SNP effect
./hiblup --snp-effect
--gebv demo.gebv.ad.txt
--bfile demo
--xrm demo.GA,demo.GD
--add --dom
--thread 32
--out demo
所有 SNP 的 SNP 效果都存储在 demo.snpeff 文件中。
注意
如果 GEBV 是使用权重 GRM 计算的,请记住提供要标记的所有 SNP 的相同权重--snp-weight。
3.17预测 GEBV/GPRS
HIBLUP 具有使用预先计算的 SNP 效应和个体水平基因型数据预测基因组估计育种值(GEBV)或基因组多基因风险评分(GPRS)的功能,这与 PLINK 软件中的功能“-score”相同,但计算速度快几倍。
进行预测的命令如下:
./hiblup --pred
--bfile demo #the binary genotype data
--score demo.snpeff #the pre-computed SNP effects
--threads 10
--out demo
请注意,基因型应该在 PLINK 二进制文件(https://http//www.hiblup.com/tutorials#genotype-file),至少5列应该包括在 SNP 效应文件(见详细格式在这里(https://http//www.hiblup.com/tutorials#snps-effect-file))。默认情况下,HIBLUP 以加性遗传效应编码基因型(即,AA Aa aa 的0 1 2) ,如果所提供的 SNP 效应都是显性的,用户可以添加一个标志“--dom”以编码显性效应的基因型(即,AA Aa aa 的 0 1 0)。但是如果在命令中同时指定了“--add”和“--dom”,那么 SNP 效果文件必须有6列,其中第5列和第6列分别是加性 SNP 效果和显性 SNP 效果。因此,如果文件中存在加性效果和显性效果,请记住在命令中添加标志“--add --dom”。
将在 work 目录中生成一个名为“ demo.bv”的文件,该文件的概述如下:
id trait1 trait2
Ind2 -0.305403 2.6644
Ind5 0.00897198 -1.36166
Ind11 0.392148 -0.653216
Ind17 0.00232218 -0.213599
Ind22 -0.359507 -2.12692
Ind45 -0.232806 0.269005
如上所示,第一列是单独的 id,其余的列是预测的 GEBV 或 GPRS,列的数量取决于 SNP 效果文件中提供的效果的数量。
3.18基因组交配
基因组交配是一种将所有可用的 SNP 联合起来的技术,用于预测雄性和雌性之间所有组合的后代的预期遗传表现(Tang,et al. 2023)。为了实现基因组交配,应该提供所有个体的基因型和所有 SNP 的加性或显性效应。SNP 效果文件的格式可以在这里找到。HIBLUP 进行基因组交配的命令如下:
./hiblup --mating
--bfile demo
--score demo.snpeff
--thread 32
--out demo
交配结果保存在 demo.Mating 文件中,这个文件的简要视图如下:
sir dam g
Ind704 Ind710 -0.0675817
Ind704 Ind711 1.06056
Ind704 Ind712 2.00997
Ind704 Ind713 -0.0879987
Ind704 Ind714 -0.492576
最后一栏是不同组合后代的预期遗传表现。
重点
(1)在 demo.bim 和 demo.snpeff 之间同名的 SNP 应该具有一致的参考基因(A1)和可选的等位基因(A2)。
(2)所有 demo.fam 的个人必须清楚标明性别(位于第5栏,1 = 男性; 2 = 女性; 其他 = 未知) ,任何性别不明的个人将被删除分析。
3.19.LD
HIBLUP 具有基于基因型等位基因计数计算成对 SNP LD 相关®的功能。命令如下:
./hiblup --ld
--bfile demo
--window-bp 1e6
--threads 16
--out demo_ldm
两个文件(**.info, *.bin)将在文件夹中生成。由于二进制文件(*.bin)不能直接打开,用户可以在上面的命令中添加一个标志--write-txt,以输出一个可读的.txt文件。
有几种设置窗口大小的选项:
--window-bp: 指定非重叠窗口(默认为1Mb,即--window-bp 1000000)的大小,其中 SNP 的数量不固定;
--window-num: 指定一个窗口中固定数量的 SNP (例如--window-num 500) ,在这种情况下窗口的大小不是常数;
--window-geno: 将整个基因组的所有 SNP 定义为一个窗口,注意如果有大量 SNP,将需要很长的时间和巨大的存储成本;
--window-file: 要指定用户预定义窗口的文本文件,请参见文件格式https://http//www.hiblup.com/tutorials#genome-windows-file。
3.20. LD成绩
LD 评分定义为 SNP 与区域内所有 SNP 之间 LD (r2)的总和,它反映了 SNP 与其他 SNP 的连锁水平,LD 评分越高表明该 SNP 与其他 SNP 相关性越高。LD 分数通常用于 LD 分数回归,以估计性状的遗传力或性状间的遗传相关。要获得单核苷酸多态性的 LD 评分,需要个体水平的基因型数据,请参阅下面的例子:
./hiblup --ldscore
--bfile demo
--window-bp 1000000
--threads 10
--out test
有几种设置窗口大小的选项:
--window-bp: 指定非重叠窗口(默认为1Mb,即--window-bp 1000000)的大小,其中 SNP 的数量不固定;
--window-num: 指定一个窗口中固定数量的 SNP (例如--window-num 500) ,在这种情况下窗口的大小不是常数;
--window-geno: 将整个基因组的所有 SNP 定义为一个窗口,注意如果有大量 SNP,将需要很长的时间和巨大的存储成本;
--window-file: 要指定用户预定义窗口的文本文件,请参见文件格式https://http//www.hiblup.com/tutorials#genome-windows-file。
将在 work 目录中生成一个名为“ test.ldsc”的文件,该文件概述如下:
id maf ldscore
M1 0.481875 1
M2 0.145 1
M3 0.320625 1
. . .
. . .
. . .
M991 0.089375 1.044
M992 0.11375 1.33231
M993 0.31875 1.2885
M994 0.103125 1
M995 0.115625 1
如上图所示,第一栏是单核苷酸多态性名称的向量,第二栏是次要等位基因频率,第三栏是计算出来的LD成绩。
用户还可以使用公开可用的 LD 分数文件来运行 HIBLUP,但有时缺少“ maf”列。由于 HIBLUP 使用“ maf”列通过计算 MAF >= 0.05的标记数来设置选项“--M”,因此,如果在运行 LD 分数回归时指定了选项“--M”(例如,文件“ XXXX.M_5_50”中的值) ,“ MAF”列将是无用的,用户可以为该列分配任何虚拟值。
3.21.LD成绩回归
LD 评分回归是总结统计中广泛使用的方法,用于估计性状的遗传力(Bulik-Sullivan,Po-Ru Loh,et al。2015)和性状之间的遗传相关性(Bulik-Sullivan,Finucane,et al。2015)。它不需要个体水平的基因型数据,仅涉及 GWAS 总结数据和参考面板计算的 LD 评分,因此与 REML 或 HE 回归相比,具有相当的时间效率和节省内存的特点。
1. 遗传力评估
./hiblup --ldreg
--sumstat demo.ma #the summary data
--lds demo.ldsc #the pre-computed LD scores
--out demo
如上文所示,应提供学习成绩摘要资料档案及学习成绩评分档案,摘要资料的档案格式及如何计算/制作学习成绩评分可参阅其他教程章节(即摘要资料及学习成绩评分)。请永远记住不要删除 LD 分数文件中的 SNP,以保持它与摘要数据文件中的一致,只要保持原样,因为用于计算这个 LD 分数的 SNP 的总数对 LD 分数回归是非常关键的。
估计的结果存储在一个名为“ demo.ldsr.h2”的文件中,该文件概述如下:
Item Intercept Intercept_SE h2 h2_SE h2_Pval
demo 1.08285 0.011433 0.122826 0.00393122 2.71554e-214
“截距”与人口结构有关,越接近1,人口分层越少。“ h2”是性状的估计遗传力,“ h2 _ Pval”是卡方检验显著性的 p 值。
2. 遗传相关性估计
遗传相关性估计的用法与遗传力估计非常相似,如果 HIBLUP 在命令中检测到多个汇总数据文件,它将自动估计遗传力和遗传相关性:
./hiblup --ldreg
--sumstat demo1.ma demo2.ma demo3.ma #the summary data of multiple traits, use space as separator
--lds demo.ldsc #the pre-computed LD scores
--out demo
请注意,性状的摘要数据的数量是不受限制的。将在工作目录中生成两个文件: 文件“ test.ldsr.h2”记录了之前描述的性状的估计遗传力; 文件“ test.ldsr.rg”存储了性状对的遗传相关性,这个文件的概述:
Item CovG CovG_SE Intercept Intercept_SE rG rG_SE rG_Pval
demo1:demo2 0.0252414 0.00716841 0.0166108 0.00819384 0.141956 0.0403148 0.000429607
demo1:demo3 0.0744506 0.00892225 0.102821 0.00671784 0.296832 0.0355727 7.15917e-17
demo2:demo3 0.262124 0.0340515 0.315166 0.0112682 0.608769 0.0790827 1.38348e-14
‘ CovG’是遗传协方差,‘ rG’是估计的性状间的遗传相关,‘ rG _ Pval’是卡方检验显著性的 p 值。
用户可指定有关的方法以进行学习能力评分回归:
--M: 在 LD 评分回归中指定 SNP 的数量。默认情况下,HIBLUP 使用 LD 评分文件中的 SNP 数量,MAF 在5% 到50% 之间,正如 Bulik-Sullivan 所建议的那样。
--chisq-max: 为拦截的第一步估计器指定 X2的最大阈值,默认值为30。
--intercept-h2: 用常数约束截距,而不是根据遗传力估计数据进行估计。
--intercept-gencov: 用常数约束截距,而不是根据遗传相关性估计数据进行估计。
3.22摘要级别的 BLUP (SBLUP)
SBLUP 模型是一种使用来自 GWAS/meta 分析的总结数据和来自具有个体水平数据的参考面板的 LD 矩阵来估计标记效应的方法(Robinson 等,2017)。摘要数据须如本文所述,以 COJO 格式制备。HIBLUP 运行 SBLUP 模型有两种方法,详情如下:
1. 使用基因型数据运行 SBLUP
运行 SBLUP 模型的第一种方法是直接使用提供的基因型数据:
./hiblup --sblup
--sumstat demo.ma #the summary data
--bfile demo
--window-bp 1e6
--h2 0.3234
--threads 16
--out demo
命令--h2是分析中性状的遗传性,如果个体水平数据可用,可以从 REML 估计性状的遗传性,或者使用汇总数据从 LD 得分回归估计性状的遗传性。
有几种设置窗口大小的选项:
--window-bp: 指定非重叠窗口(默认为1Mb,即--window-bp 1000000)的大小,其中 SNP 的数量不固定;
--window-num: 指定一个窗口中固定数量的 SNP (例如--window-num 500) ,在这种情况下窗口的大小不是常数;
--window-geno: 将整个基因组的所有 SNP 定义为一个窗口,注意如果有大量 SNP,将需要很长的时间和巨大的存储成本;
--window-file: 要指定用户预定义窗口的文本文件,请参见文件格式https://http//www.hiblup.com/tutorials#genome-windows-file。
如果定义的窗口大小中 SNP 的数量非常大(例如,超过10k) ,建议添加 Flag--pcg 来快速计算分析中的 SNP 效应。
2. 使用预先计算的 LD 相关矩阵运行 SBLUP
与使用基因型数据不同,使用 LD 相关矩阵来拟合 SBLUP 模型更为直接。尽管这种策略比第一种更有计算效率和节省内存,但应该指出的是,所有的 SNP 都应该满足 Hardy-Weinberg 均衡。如果没有,估计的 SNP 效应将是有偏差的,导致不良的预测性能。
./hiblup --sblup
--sumstat demo.ma #the summary data
--ldm demo_ldm #the pre-computed LD correlation matrix
--h2 0.3234
--threads 10
--out demo
命令--ldm 用于指定 LD 相关矩阵,该矩阵可以由 HIBLUP 使用个体水平的基因型数据来计算,请参阅此处的更多细节。此外,如果一个窗口中的 SNP 数量相当大(例如,超过10k) ,建议添加 Flag--pcg,以便在分析中快速计算 SNP 效应。
成功运行后,将在 work 目录中生成一个名为“ demo.snpeff”的文件,如下所示:
id a1 a2 freq_a1 demo
M1 A G 0.1285 -0.000963937
M2 T C 0.1285 -0.00108931
M3 A G 0.1062 0.00588629
M4 G A 0.1285 -0.00164344
M5 A C 0.2459 -0.00100206
如上所示,最后一列列出了估计的 SNP 效应。为了获得个体的 GEBV 或 GPRS 预测值,我们建议使用 HIBLUP 来实现它(参见此处) ,因为我们测试了它比 PLINK 中的“—— score”函数快几倍。
3.23多性状 SBLUP (MT-SBLUP)
多性状 SBLUP 模型是从单性状 SBLUP 模型扩展而来的,在预测精度上优于单性状 SBLUP 模型(Robert,Zhhong,et al. 2018)。有两种方法可以拟合 MT-SBLUP 模型,通过个体基因型数据或 LD 相关矩阵,见更多关于 SBLUP 模型章节的细节。在这里,我们只展示了如何使用预计算的 LD 相关矩阵来拟合 MT-SBLUP 的示例:
./hiblup --sblup
--sumstat demo1.ma demo2.ma demo3.ma demo4.ma #the summary data, use space as separator
--ldm demo_ldm #the pre-computed LD correlation matrix
--h2 0.3234 0.1256 0.6345 0.3536
--rg 0.1336 0.5567 0.2345 0.8454 0.3446 0.4633
# --pcg #use PCG for fast computing
--threads 10
--out demo
注意,分析中的性状摘要数据的数量是不受限制的。摘要数据应以 COJO 格式准备,如下所述,LD 相关矩阵存储在二进制文件中,可由 HIBLUP 输出(见此处)。必须指定性状的遗传力和性状间的遗传相关性,如果个体水平数据可用,可以通过 REML 估计这些遗传参数,或者通过使用总结数据的 LD 评分回归估计这些遗传参数。遗传相关性的输入顺序为矩阵的下三角形(以上述命令为例,输入顺序应为: 1-21-31-42-32-43-4)。
成功运行后,将在 work 目录中生成一个名为“ demo.snpeff”的文件,如下所示:
id a1 a2 freq_a1 demo1 demo2 demo3 demo4
M1 A G 0.1285 -0.000963937 -0.000577569 -0.000792698 0.000175215
M2 T C 0.1285 -0.00108931 -0.000597102 -0.000825137 0.000177501
M3 A G 0.1062 0.00588629 0.00155157 0.00270818 0.000154987
M4 G A 0.1285 -0.00164344 -0.000557257 -0.000874613 0.000155528
M5 A C 0.2459 -0.00100206 -0.000456737 -0.000855748 -0.000422206
如上所示,估计的 SNP 效应在最后几列中按性状列出。为了获得个体的 GEBV 或 GPRS 预测值,我们建议使用 HIBLUP 来实现它(参见此处) ,因为我们测试了它比 PLINK 中的“—— score”函数快几倍。















 2406
2406











 被折叠的 条评论
为什么被折叠?
被折叠的 条评论
为什么被折叠?
 到【灌水乐园】发言
到【灌水乐园】发言









