







下载序列简单不过,无非就是联网NCBI主页,选择数据库后输入AC号或GI号后直接下载。但是如何大批量下载,而且下载的序列是指定的AC或GI的呢?实现这一目的通常办法是借助一些生物软件的检索功能,诸如:Bioedit、Geneious、MacVector等。其实,NCBI自带的Batch Entrez 只需简单三步即可轻松完成这一任务。
【准备工作】
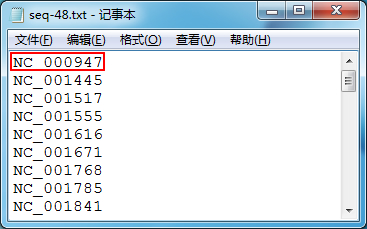
创建一个需要下载序列AC号的列表文件,每行一个独立的AC号,保存为文本文件:
【简要流程】
1、打开网页,粘贴这个网
址:http://www.ncbi.nlm.nih.gov/sites/batchentrez?
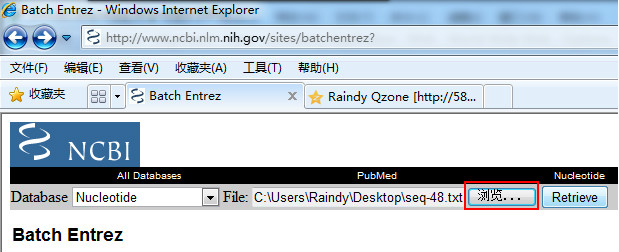
点击“浏览”按钮,选择事先准备带AC号的文本文件后,点击“Retrieve”开始检索,数秒后即可返回检索的序列记录;
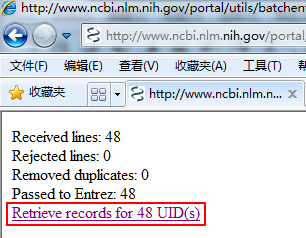
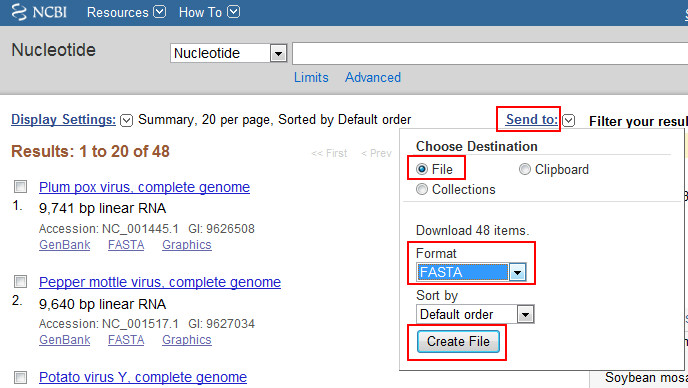
3、点击检索到的序列记录“Retrieve records for 48 UID(s)”即可,后面就是“Send to” 保存要下载的序列,后面的操作就不在赘述,详见前面一些相关教程。

















 8589
8589

 被折叠的 条评论
为什么被折叠?
被折叠的 条评论
为什么被折叠?
 到【灌水乐园】发言
到【灌水乐园】发言









