







什么是全基因组关联分析?(Genome-Wide Association Study,GWAS)
全基因组关联分析(GWAS)是一种在全基因组范围内搜索遗传变异(通常是单核苷酸多态性,SNP)与复杂性状之间关联的方法。
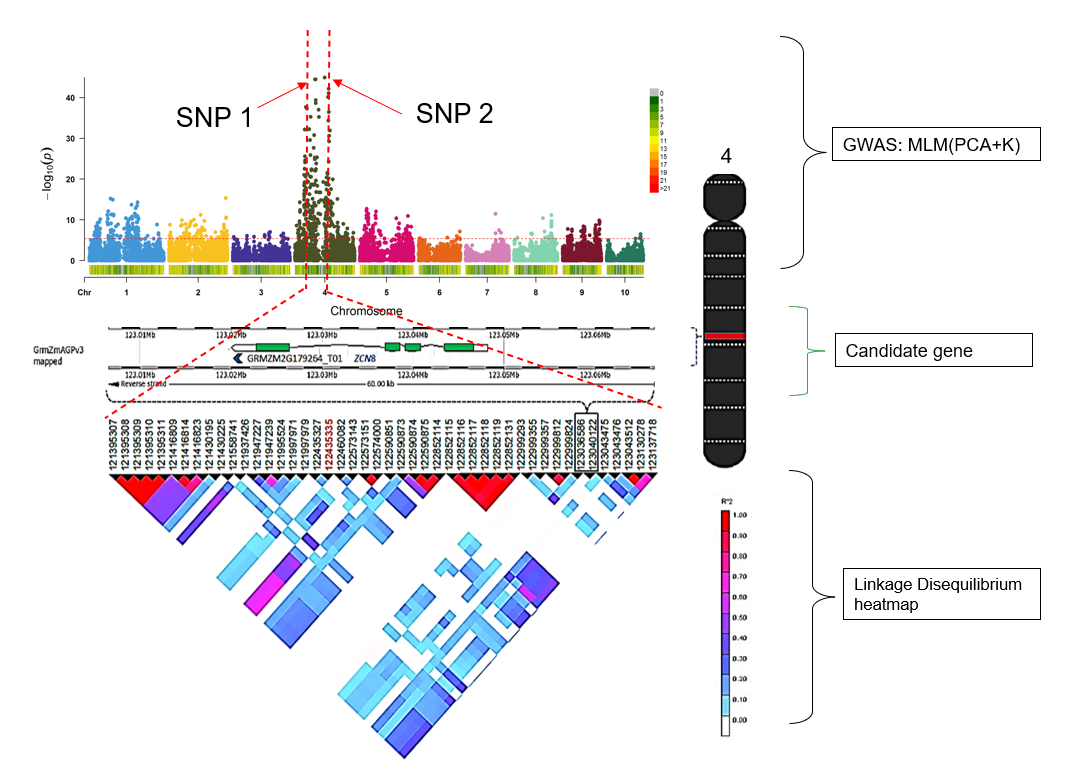
其核心思想是通过比较群体中不同个体的基因型与表型,识别出与目标性状显著相关的基因位点。GWAS基于连锁不平衡(LD)的原理,即相邻的遗传变异倾向于一起遗传,因此可以通过标记SNP间接捕获致病变异。
GWAS的应用场景
-
复杂性状解析:用于揭示控制复杂农艺性状(如产量、品质、抗病性等)的遗传基础,探索遗传学机理。
-
疾病抗性研究:发现与植物病害抗性相关的基因,为育种提供候选基因,其他性状也以此类推。
-
分子育种:辅助分子标记辅助选择(MAS)和基因组选择(GS)。
数据分析过程与原理
表型数据收集:精确、可靠的表型测定是关键。需在多环境、多重复下评估表型特征等数据,以减少环境误差。
基因型数据获取:利用SNP芯片或高通量测序技术获取全基因组SNP数据。
数据质量控制(QC)
- 标记过滤:删除缺失率高、次等位基因频率(MAF)低、偏离哈迪-温伯格平衡的SNP。
- 个体过滤:剔除基因型缺失率高或有杂合度异常的个体。
群体结构和亲缘关系分析
- 主成分分析(PCA):识别和校正群体结构。
- STRUCTURE或ADMIXTURE分析:确定群体的组分。
- 亲缘关系矩阵(Kinship Matrix):估计个体间的亲缘关系。
关联分析模型构建
一般线性模型(GLM):y=Xβ+ϵ
- y:表型值向量
- X:基因型矩阵
 最低0.47元/天 解锁文章
最低0.47元/天 解锁文章



















 5万+
5万+

 被折叠的 条评论
为什么被折叠?
被折叠的 条评论
为什么被折叠?
 到【灌水乐园】发言
到【灌水乐园】发言











