







 本文介绍五种在R中进行基因ID类型转换的方法,包括使用clusterProfiler、AnnotationDbi等包进行ENTREZID到ENSEMBL、SYMBOL等类型的转换,适用于生物信息学研究。
本文介绍五种在R中进行基因ID类型转换的方法,包括使用clusterProfiler、AnnotationDbi等包进行ENTREZID到ENSEMBL、SYMBOL等类型的转换,适用于生物信息学研究。
对于这四种方法的使用参数在代码里面写了,你可以看看
先把基因集对应的注释包安装了,并且加载出来
查找对应的注释包请看:https://blog.csdn.net/weixin_40739969/article/details/103186027
library(org.Hs.eg.db)#这个是属于注释包,每个基因集可能对应的注释包不一样,要从基因集所在的平台找到对应的注释包,然后从bioconductor获取安装。
基因集准备
data(geneList, package="DOSE")
gene <- names(geneList)[abs(geneList) > 2]
head(gene)
结果是
'4312' '8318' '10874' '55143' '55388' '991'
第一种方法
安装clusterProfiler包,bitr转换ID函数属于这个包
source("http://bioconductor.org/biocLite.R")
options(BioC_mirror="http://mirrors.ustc.edu.cn/bioc/")
biocLite("clusterProfiler")
library("clusterProfiler")
gene.df <- bitr(gene, fromType = "ENTREZID", #fromType是指你的数据ID类型是属于哪一类的
toType = c("ENSEMBL", "SYMBOL"), #toType是指你要转换成哪种ID类型,可以写多种,也可以只写一种
OrgDb = org.Hs.eg.db)#Orgdb是指对应的注释包是哪个
head(gene.df)
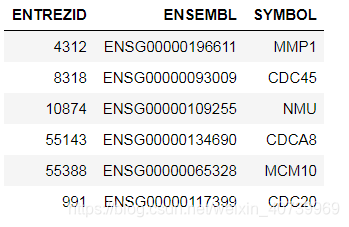
第二种方法
library(AnnotationDbi)
mySymbols <- mget(gene,
org.Hs.egSYMBOL, #这个是可以选择的,选择不同,转换的ID类型也不一样
ifnotfound=NA)
转换成Symbol ID
head(mySymbols)
class(mySymbols)

第三种方法
geneIDselect <-select(org.Hs.eg.db, #.db是这个芯片数据对应的注释包
keys=gene,
columns=c("SYMBOL","ENSEMBL","GENENAME"), #clolumns参数是你要转换的ID类型是什么,这里选择三个。
keytype="ENTREZID" )#函数里面的keytype与keys参数是对应的,keys是你输入的那些数据,keytype是指这些数据是属于什么类型的数据。
head(geneIDselect)
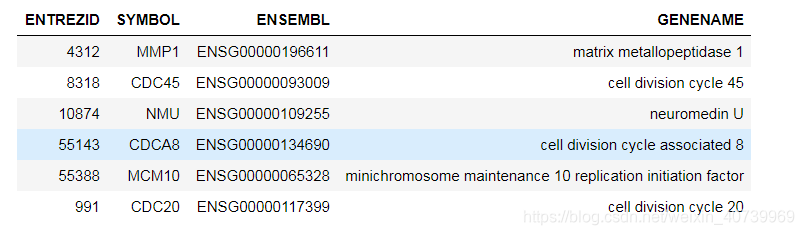
第四种方法
gene <-mapIds(org.Hs.eg.db, #.db是这个芯片数据对应的注释包
keys=gene,
column="SYMBOL", #clolumns参数是你要转换的ID类型是什么,只能选择一个。
keytype="ENTREZID" )#函数里面的keytype与keys参数是对应的,keys是你输入的那些数据,keytype是指这些数据是属于什么类型的数据。
head(geneIDselect)

第5种方法
annotate::getEG() #有空再写吧

















 3581
3581

 被折叠的 条评论
为什么被折叠?
被折叠的 条评论
为什么被折叠?
 到【灌水乐园】发言
到【灌水乐园】发言









