







涉及到gene的传统分析环节,就是表达量矩阵数据处理,最频繁的就是传统的表达量芯片的差异分析和富集分析;
这些分析都是基于基因的,而基因有多种多样的id体系,而且不同的数据分析环节经常是需要进行id的转换!
1,不同数据库的gene的id体系:
在基因组学和分子生物学研究中,基因的标识符是理解和交流基因信息的关键。关于基因的symbol、人类基因命名委员会(HGNC)、Entrez ID和Ensembl ID的解释:

理解这些标识符的重要性:
标准化:基因符号和ID提供了一种标准化的方式来引用基因,这对于科学出版物和数据库的一致性至关重要。
数据检索:这些标识符是检索基因相关信息的关键,如序列数据、表达模式、功能注释等。
跨数据库兼容性:不同的数据库可能使用不同的标识符系统,但它们通常可以相互转换,以便于数据的整合和分析。
在实际研究中,研究人员可能会根据需要使用这些不同的标识符来查找特定基因的信息,或者在比较不同研究结果时进行基因标识符的转换。
暂时没有统一的命名体系的基因
基因命名和标识符系统是多样化的,反映了基因的不同类型和功能 :

2,如何转换gene id:
(1)在线网页转换工具(比较权威的生物数据库):
①bioDBnet(Biological DataBase network,生物数据库网络)
https://biodbnet-abcc.ncifcrf.gov/db/db2db.php

参考https://mp.weixin.qq.com/s/VZ3gh-DlB0MqUfTpmRG80A
②Ensembl数据库是涵盖Human、Mouse、Zebrafish等模式动物,拟南芥、水稻等模式植物,真菌、原生动物、后生动物、细菌的基因组数据库。
https://asia.ensembl.org/index.html

参考https://mp.weixin.qq.com/s/VZ3gh-DlB0MqUfTpmRG80A
③DAVID(The Database for Annotation, Visualization and Integrated Discover)是一款整合了生物学数据和分析工具的生物信息数据库,为基因或蛋白提供系统的生物学注释信息。
https://david.ncifcrf.gov/home.jsp

参考https://mp.weixin.qq.com/s/VZ3gh-DlB0MqUfTpmRG80A
④g:Profiler中的g:convert可以实现各种基因,蛋白质,微阵列探针和许多其他类型的ID之间进行转换。涵盖60多个物种、至少40多种ID类型。
https://biit.cs.ut.ee/gprofiler/convert

参考https://mp.weixin.qq.com/s/VZ3gh-DlB0MqUfTpmRG80A
⑤KEGG(Kyoto Encyclopedia of Genes and Genomes)是一个整合了基因组、化学和系统功能信息的数据库。
https://www.kegg.jp/kegg/tool/conv_id.html

参考https://mp.weixin.qq.com/s/VZ3gh-DlB0MqUfTpmRG80A
⑥Uniprot(Universal Protein)是信息最丰富、资源最广的蛋白质数据库。整合Swiss-Prot、 TrEMBL 和 PIR-PSD 三大数据库。
https://www.uniprot.org/id-mapping

参考https://mp.weixin.qq.com/s/VZ3gh-DlB0MqUfTpmRG80A
⑦metascape:
https://metascape.org/gp/index.html#/main/step1
(2)使用R包进行gene id转换:
参考https://mp.weixin.qq.com/s/2Z2qMnak7F2G_bo-9YbNxw
https://mp.weixin.qq.com/s/Ba-KJc2_6PmEj-KtcQ9CGQ
https://blog.csdn.net/weixin_40739969/article/details/89354167
https://www.biostars.org/p/22/
①bioMart:除了上面的1个在线ensembl数据库,还有一个R包——biomaRt:
参考https://mp.weixin.qq.com/s/mb4rXd_sW0toPbGrJRs-Jg
②clusterProfiler::bitr:也就是bitr包:
参考https://mp.weixin.qq.com/s/mb4rXd_sW0toPbGrJRs-Jg
③Genekitr:
参考https://www.genekitr.fun/gene-id-conversion-1
④AnnotationDbi::mapIds:
参考https://mp.weixin.qq.com/s/KsZGc4m8QXEZgKZbuSMhjg
⑤gprofiler:也有R包,和前面一样还有1个在线网站
(3)自己写脚本:
参考https://mp.weixin.qq.com/s/2Z2qMnak7F2G_bo-9YbNxw
①use Web Crawler:

参考https://mp.weixin.qq.com/s/5u_7wJ1raSrltzum4RbSQA
②获取gtf/gff注释文件,然后自己写linux脚本(awk、sed、grep等3剑客)
参考https://mp.weixin.qq.com/s/rKErPfTCFfcVJX_laGfpmg

(4)最常见的gene id转换:
ENSEMBL、SYMBOL、ENTREZID,这3种格式之间的转换
参考:
https://mp.weixin.qq.com/s/mBNiTFKhGmLtMLWkrqLD8g
3,id转换过程中会遇到的问题:
(1)转换数据不全:通常是转换之后id大幅损失(id匹配率低下问题)
建议优先用ensembl+bioMart
参考https://mp.weixin.qq.com/s/kw2x2Tku53SiGeFAMoAwaw
http://www.bio-info-trainee.com/8032.html
参考数据库、R包等都会影响
org.Hs.eg.db一般会偏少,
应对策略:
①使用biomart多点
②使用genecode数据库的gtf注释,也就是从gtf上获取注释(但是注意哟啊使用匹配的gtf版本信息!!!)
③TCGA的gene id转换问题可以去ucsc的xena浏览器里下载几乎完美匹配的id

④如果用bitr少用org.Hs.eg.db数据库等
⑤多用Ensembl ID
⑥参考https://github.com/ixxmu/mp_duty/issues/1014
https://mp.weixin.qq.com/s/evF9kFNcjcJYYDB4vgTbow
R包影响比参考数据库影响比较小



既然用ensembl多,那就用ensembl自家的biomaRT包,即3.3法推荐常用!!!
其他方法补充https://github.com/paulgeeleher/pRRophetic2/blob/master/pRRophetic/vignettes/prepareData.R




(2)要多少的匹配率才合适?

参考https://bmcbioinformatics.biomedcentral.com/articles/10.1186/1471-2105-13-229

参考:https://www.ncbi.nlm.nih.gov/pmc/articles/PMC1779800/

这个问题到github issue、google、以及一些转换R包的issue、research gate、biostars上都没怎么找到,还找了一些生信英文期刊;
最后终于找到这么一点:
https://www.researchgate.net/figure/Entrez-ID-to-gene-symbol-conversion-accuracy_tbl1_230831496
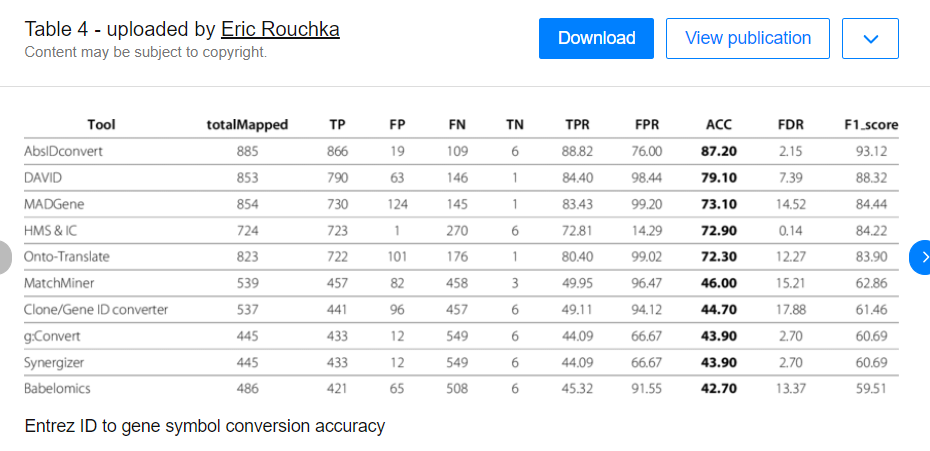
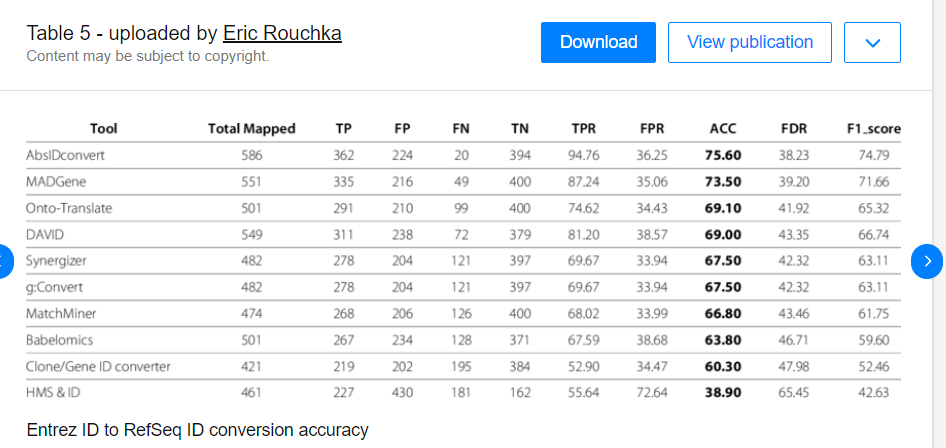
从上面的table来看,个人认为最好是80%以上,当然越多越好!
参考文献:https://bmcbioinformatics.biomedcentral.com/articles/10.1186/1471-2105-13-229

(3)一个基因有两个id等奇怪问题:
参考:http://www.bio-info-trainee.com/8914.html

id的重复值问题:

(4)尽量参考高质量的生信创作者:
生信技能树、生信菜鸟团、生信益站、生信宝典、生信星球+一些生物技术分析公司公众号,其他的个人博客少看、看多了误导性很强

















 2444
2444

 被折叠的 条评论
为什么被折叠?
被折叠的 条评论
为什么被折叠?
 到【灌水乐园】发言
到【灌水乐园】发言









