







AMBER分子动力学模拟之TOP准备-- HIV蛋白酶-抑制剂复合物(1)
我们以HIV蛋白酶-抑制剂复合物为例子,跑Amber动力学模拟
下载1phv
从PBD下载文件:https://www.rcsb.org/

PDB文件预处理
我们以 “protein+(water) + ligand=complex” 为例来说一下如何处理:
将1HPV.pdb文件保存为三个文件; protein.pdb ligand.pdb complex.pdb (只保留原子坐标信息即可)
[Luos@node88 1hpv] $ cp 1hpv.pdb complex.pdb
[luos@node88 1hpv]$ cp 1hpv.pdb ligand.pdb
[Luos@node88 1hpv]$ cp 1hpv.pdb protein.pdb
protein.pdb只保留蛋白三维信息
ligand.pdb只保留分子三维信息
complex.pdb 只保留蛋白和分子三维信息
ligand.pdb如下信息:

protein.pdb如下信息:

complex.pdb可以将ligand.pdb和protein.pdb合并
加氢原子
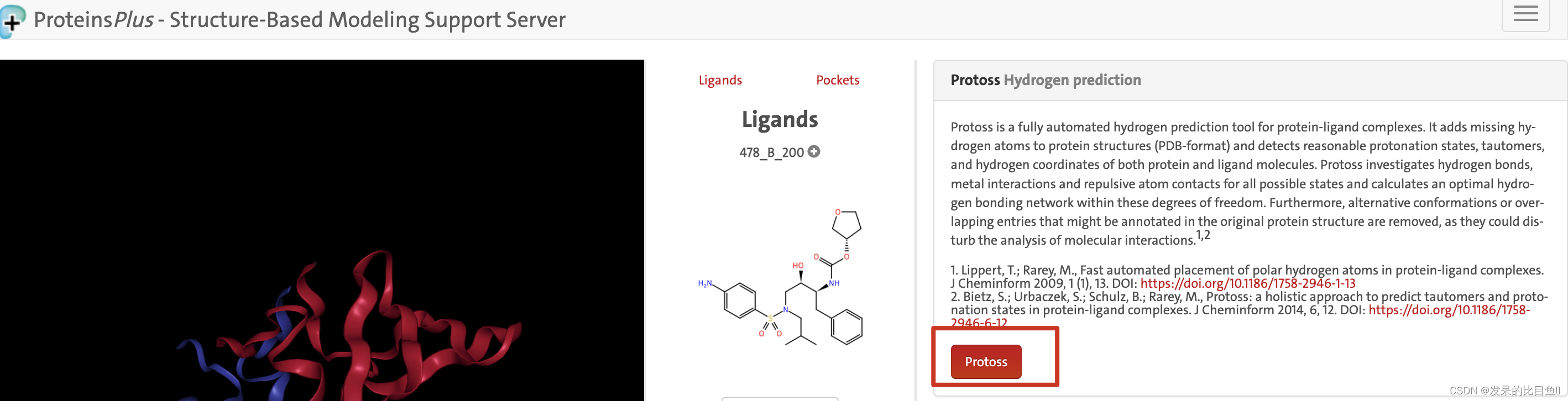
对小分子的加氢处理需要用到其他额外的软件, 如Schrodinger, DiscovereyStudio等软件, 加氢的目的是因为晶体结构一般是无法解 析氢原子的, X-ray不能解析氢原子, NMR可以。对于加氢这个处 理过程我们一般都用软件进行加氢, 而不用AMBER自带的加氢程 序。这里推荐使用protein+ (https://proteins.plus/), 不同的软件加氢 结果可能不会完全相同, 目前尚不存在一个完全可靠的加氢软件, 加氢后的文件中小分子中的化学键(单双建、苯环、…)、分子 式…需要我们核对一下, 这一个是经验性的检查。
其他加氢方式:蛋白质加氢
蛋白加氢
我们可以使用PDB网站的ID去加氢,注意区分大小写

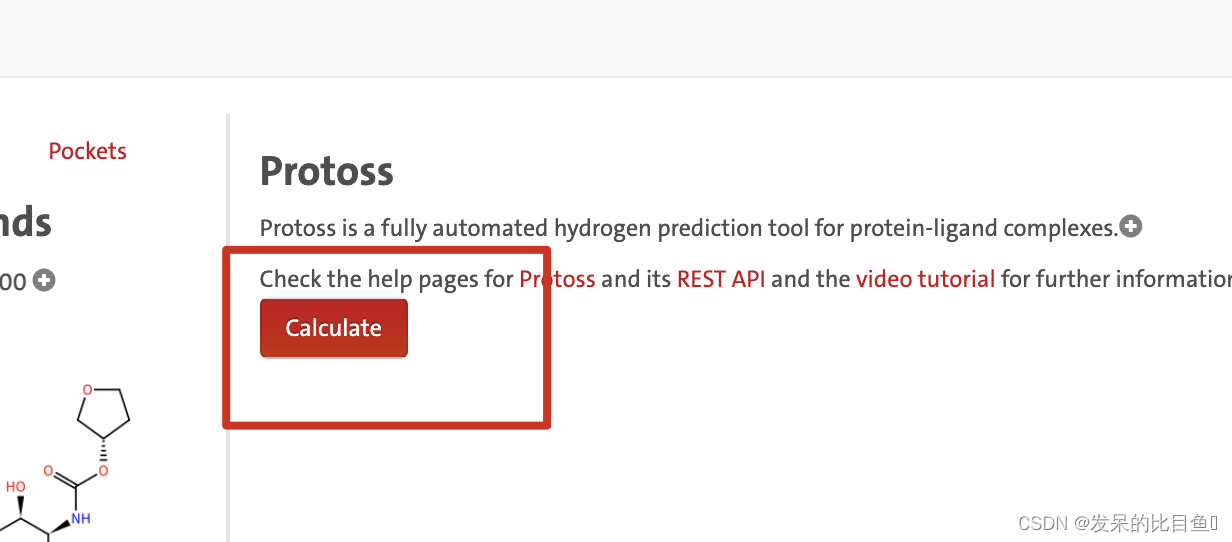

注意:下载后的文件需要校验一下(去除水分子),只保留坐标信息。
最后文件:protein_h.pdb, ligand_h.pdb
小分子生成电荷处理
生成小分子的电荷文件
antechamber -i ligand_h.pdb -fi pdb -o ligand.mol2 -fo mol2 -rn MOL -c bcc -nc 0 -pf y &
上面命令解释如下:
-ilig_h.pdb-fi pdb: -i, 参数i, 表明输入(input)【下同】; ligand_h.pdb, 输入的文件; -fi, 参数f,文件(file)格式, pdb, 表示输入的格式为 p d b \mathrm{pdb} pdb 。(注:该pdb文件必须包含详细的成键信 息)-oligand1.mol2 -fo mol2: -o, 参数o, 表明输入(output)【下同】; ligand.mol2, 输出的文件; m o l 2 \mathrm{mol} 2 mol2,规定输出文件的格式为 m o l 2 \mathrm{mol} 2 mol2 。-cbcc:采用的方法为bcc。-nc− n c -\mathrm{nc} −nc, 电荷的数量(number of charge): 0 。(根据各自体系填写)
&:后台运行符
完成后会产生ligand.mol2文件。
对于如何查看带什么电荷,下面网站可以作为参考:
https://xundrug.cn/molgpka

用parmchk模块生成小分子的键长、键角和二面角。
parmchk2 -i ligand.mol2 -f mol2 -o ligand. frcmod
-i ligand.mol2 -f mol2输入 mol2文件;
-o ligand.frcmod: 输出键长、键角和二面角frcmod文件。

对蛋白PDB文件检查
查看所挑选的肽链是否断链;
打开原始pdb文件 vi hpv.pdb
检索关键词MISS查看有没有断链

查看是否存在肽链是否存在二硫键;
继续检索SSBOND关键词, 查看二硫键情况

查看是否出现非标准残基或同构情况。
检查ATOM列是否一致, 残基名称前是否出现
A
/
B
+
\mathrm{A} / \mathrm{B}+
A/B+ 残基名的情况, 若有, 保留
A
\mathrm{A}
A 行, 删除
A
\mathrm{A}
A 字符, 使第四列对齐, 并且删除
B
\mathrm{B}
B 行。
Leap蛋白分子处理
用Leap模块对小分子和蛋白质进行预处理 若无硫键键, 对蛋白质-配体leap处理。
vim leap.in
写入
source leaprc.protein.ff14SB # 蛋白立场
source leaprc.water.tip3p # 水力场
source leaprc.gaff
loadamberparams ligand.frcmod # 加载分子力场文件
loadamberparams frcmod.ionsjc_tip3p #离子力场
p = loadpdb protein.pdb
l = loadmol2 ligand.mol2
c = combine {p l} ## 合并蛋白和分子
savepdb p pro.pdb
savepdb l lig.pdb ## 保存pdb格式的文件
savepdb c com.pdb
saveamberparm p pro.top pro.crd ## 保存成拓扑和原子坐标文件, prmtop文件同时包含了分子的力场参数信息和拓扑信息,拓扑信息指分子的键连信息
saveamberparm l lig.top lig.crd
saveamberparm c com.top com.crd
solvateoct c TIP3PBOX 12.0 ## 于复合物中添加八面体水盒子,截止距离为12埃
charge c ##添加无机盐离子。中和电荷
addions c Cl- 0
addions c Na+ 0
savepdb c pep.pdb ## 保存处理好的pdb文件
saveamberparm c pep.top pep.crd
quit
利用Leap模块进行体系的预处理, 生成需要的参数文件
tleap -sf leap.in
可以看到leap模块正常完成, 没有出现error 具体的详细信息我们可以查看leap.log文件 vi leap.log

start;
注意,如果pdb蛋白分子有二硫键, leap.in写入文件就不一样。
针对有二硫键的蛋白体系, 比如1NPO.pdb
第一步 vi 打开1nop.pdb 搜索关键词SSBOND,确实存在二硫键

第二步, 命令: pdb4amber -i 1npo.pdb -o protein.pdb -d -y 得到这些文件

第三步,打开protein_sslink文件,如下:

第四步,打开protein.pdb文件,检查对应的CYS残基是否已经改为CYX ( ambber自动改残基名字) 或者 grep CYX protein.pdb
二硫键在amber中名为CYX ,把对应的 CYS改为CYX。
第五步,打开protein.pdb 把文件末尾所有的 CONECT 行删除

第六步,在tleap 输入文件中加入二硫键信息

最后一步, 生成参数, 与无二硫键方式一样
tleap - f tleap.in
end;
最后的TOP文件已经准备好了,下一步我们开始做模拟
















 501
501











 被折叠的 条评论
为什么被折叠?
被折叠的 条评论
为什么被折叠?
 到【灌水乐园】发言
到【灌水乐园】发言











