






组会上老师提了一嘴,测序的公司不做这个分析,但我觉得这应该用RNAseq数据就能做吧?最近刚好在搞RNAseq标准分析,顺便学个高级的。
文章目录
各种可变剪接软件
搜了一圈发现好几个软件,选了可视化最好看的miso,但是做到一半没做出来,最后用了和miso效果差不多的rmats。先贴上各个软件的主页:
SUPPA2,DEXSeq,SGSeq,LeafCutter ,SpliceGrapher ,MISO,rMATS + rmats2sashimiplot
SUPPA2
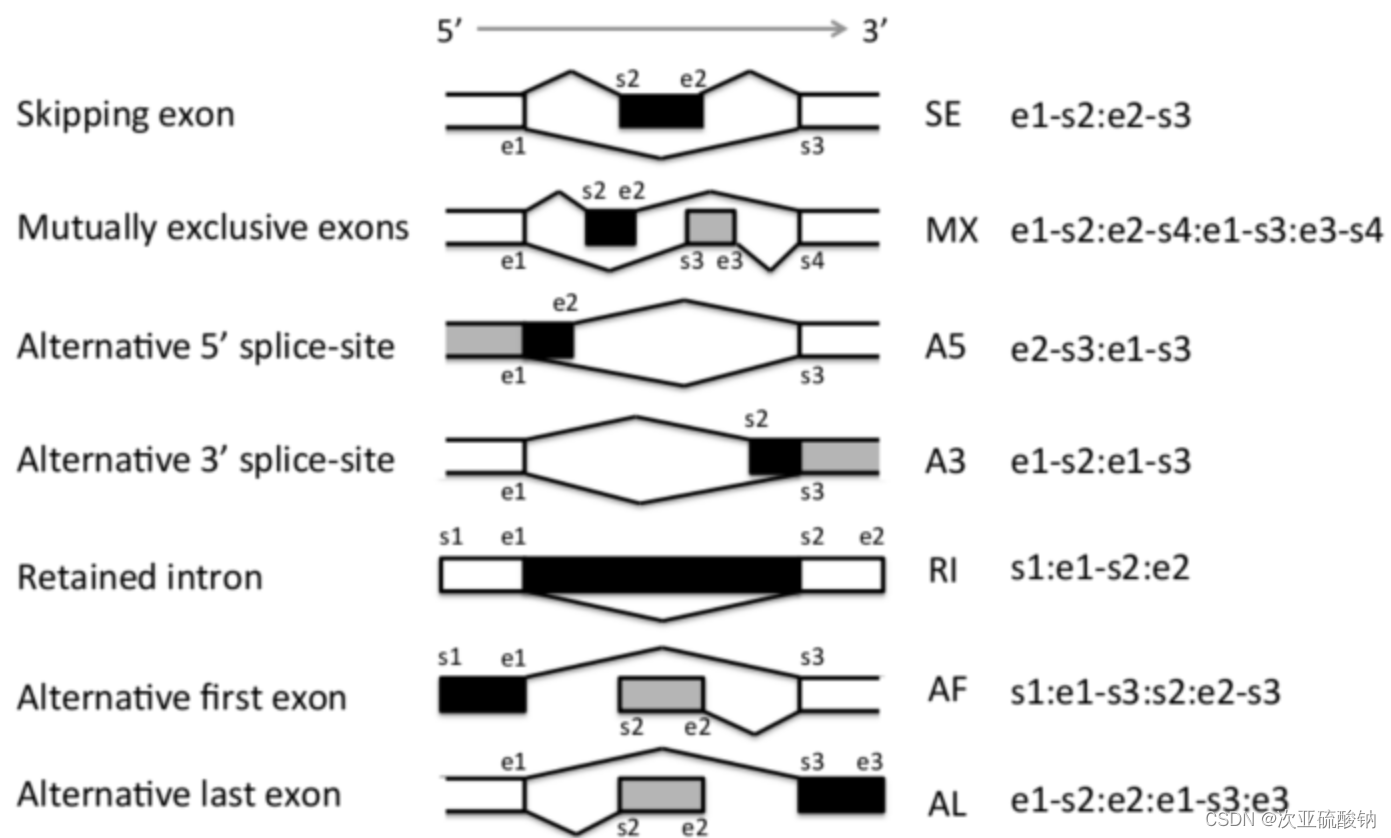
能够识别7总可变剪接事件:
- 外显子跳跃(ES)
- 3’端可变剪接(A3)
- 5’端可变剪接(A5)
- 第一外显子替换(AF)
- 末尾外显子替换(AL)
- 外显子互斥(ME)
- 内含子保留(RI)
输出文件是这样的:
event_id cluster_id cond1_PSI_avg cond2_PSI_avg
ENSG00000004897;SE:chr17:45247408-45249283:45249430-45258928:- 1 0.967582617827 0.885135296685
ENSG00000004961;A5:chrX:11129580-11130139:11129543-11130139:+ 1 0.950872555163 0.720740016826
ENSG00000005189;A3:chr16:20824624-20826249:20824624-20826307:+ 0 0.632147193342 0.332088364504
ENSG00000005189;A5:chr16:20818139-20818274:20818027-20818274:+ 0 0.344510537149 0.161862745525
ENSG00000005189;SE:chr16:20851790-20855256:20855348-20855951:+ -1 0.853283401943 0.548674896371
ENSG00000005194;SE:chr16:57463192-57464170:57464251-57466399:- -1 0.971516032983 0.829712829609
没有自带的可视化方法,只能通过查找差异大的结果用IGV可视化,这效果一言难尽😂
参考:
https://github.com/comprna/SUPPA
https://cloud.tencent.com/developer/article/1892323#3.8
DEXSeq
这个软件是以counts矩阵作为输入,并且要提供实验设计表,比较特别。可视化结果稍微好看点,但还是不太行
参考:
http://www.bio-info-trainee.com/bioconductor_China/software/DEXSeq.html
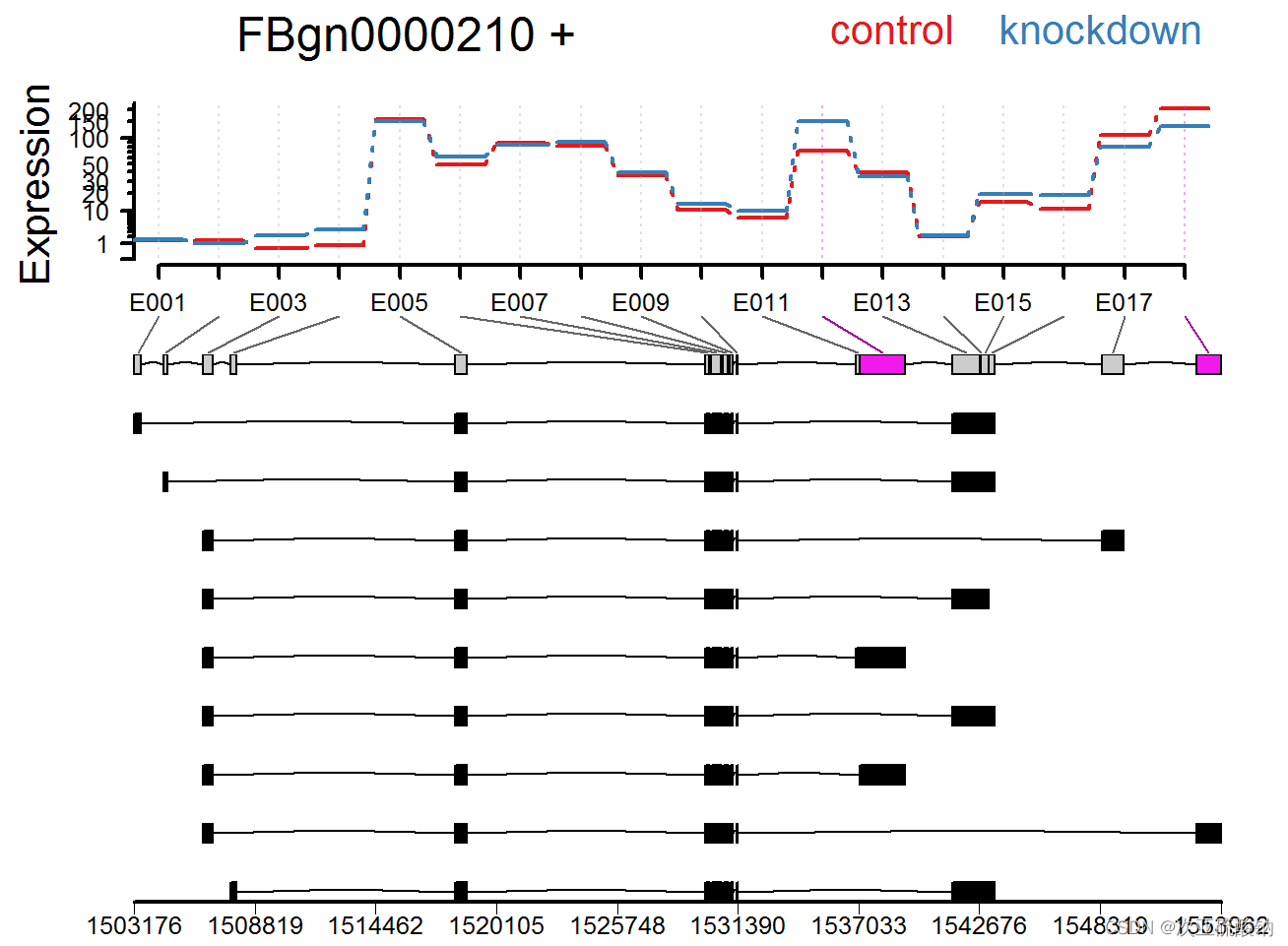
SGSeq
全局查看存在的可变剪接事件,并可视化:
挑选其中一个基因可视化其差异表达情况:
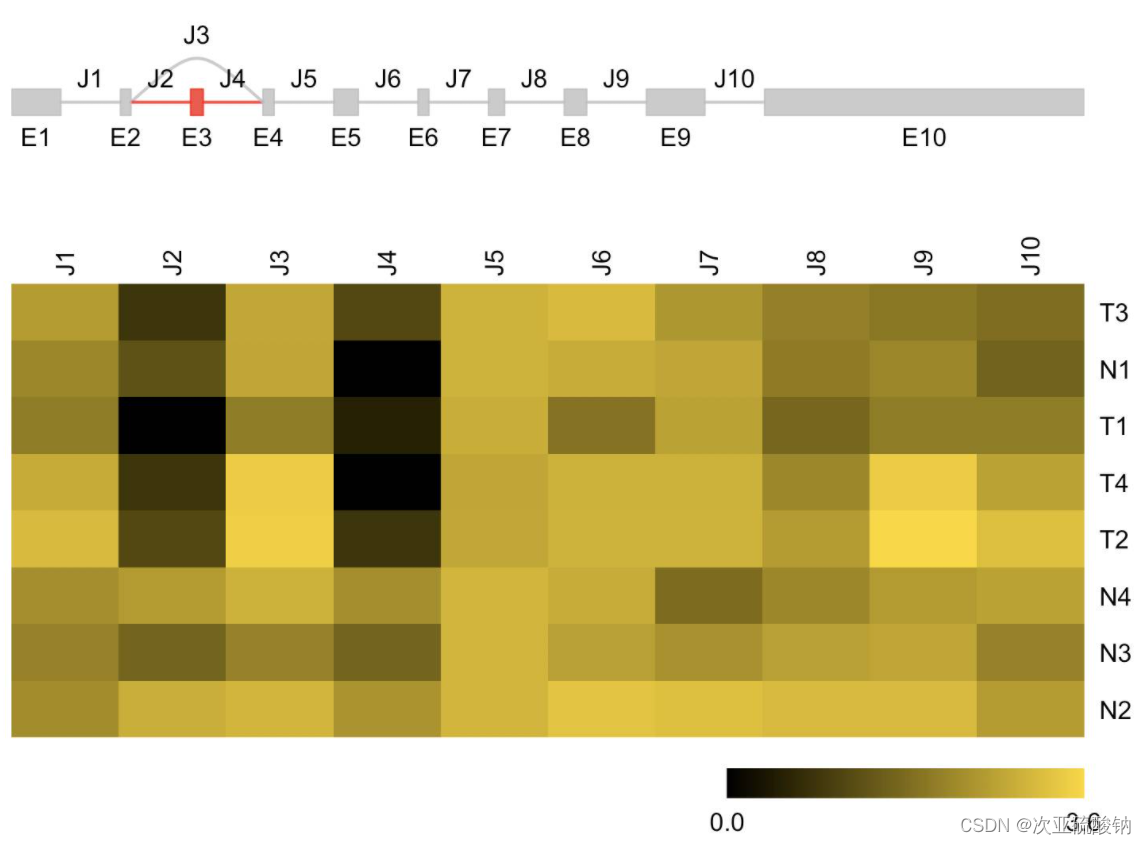
用bam和注释文件输入就可以,看着运行也不难,具体到基因的可视化效果不错!值得一试。
参考:
https://mp.weixin.qq.com/s?__biz=MzAxMDkxODM1Ng==&mid=2247485574&idx=1&sn=c9b36684206b52428d5d3bfcfddd765c&scene=21#wechat_redirect
LeafCutter
咋看一下,好像是挺麻烦的,而且看到这自带的可视化效果,就不想了解了😂(好像可以用其他方法可视化)
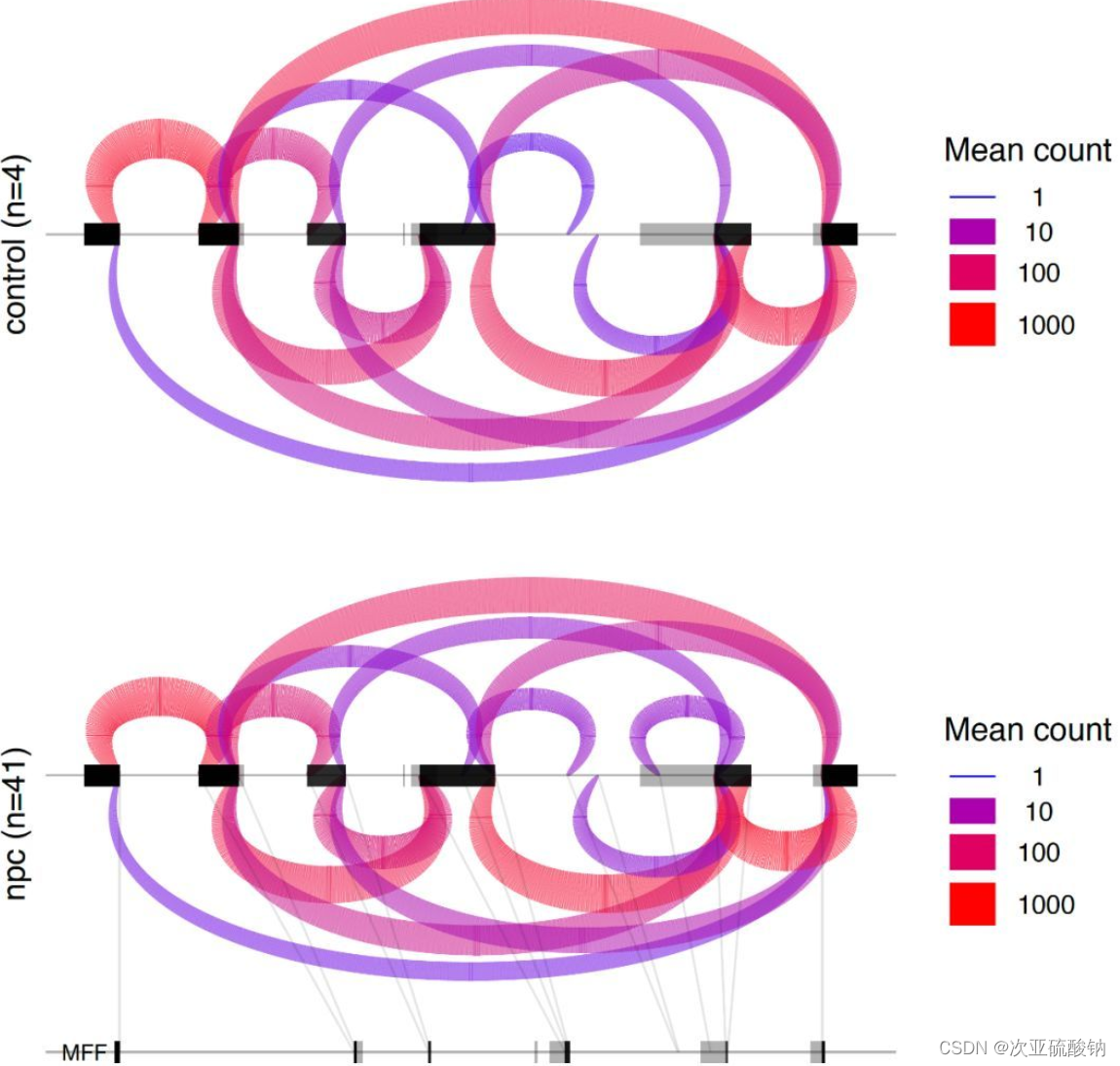
参考:
https://cloud.tencent.com/developer/article/1539862?from=article.detail.1625308
SpliceGrapher
这个软件用的sam文件作为输入,使用起来比较复杂,而且比较老(2012-13年发布),但是做的图还不错。
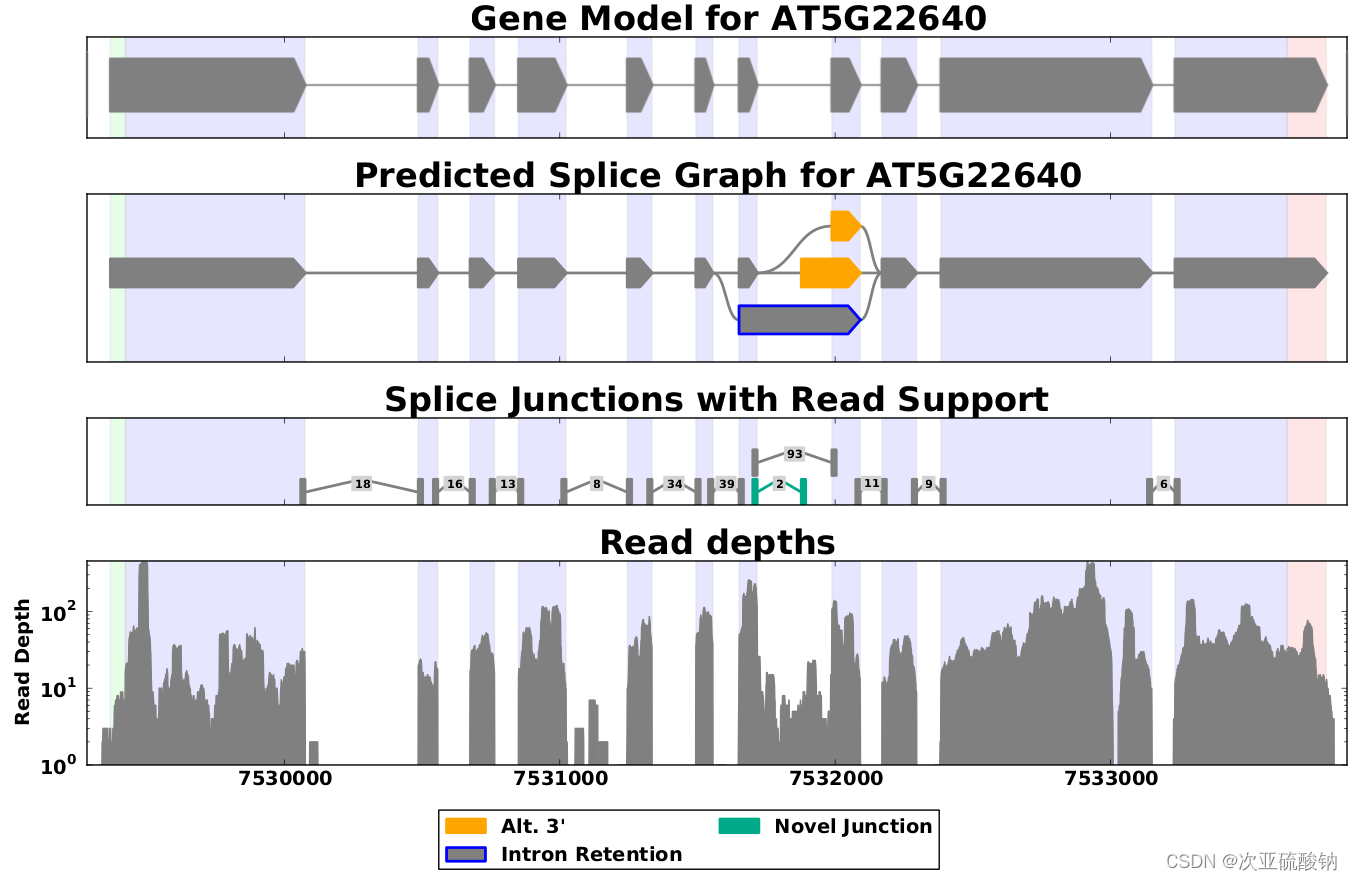
参考:
http://splicegrapher.sourceforge.net/
http://www.chenlianfu.com/?p=2290
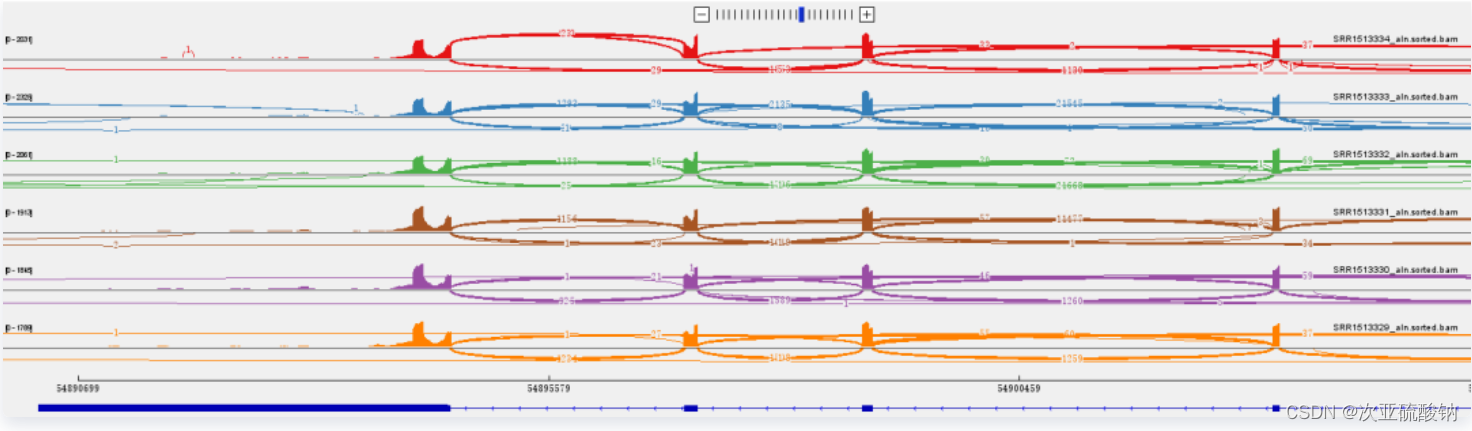
MISO
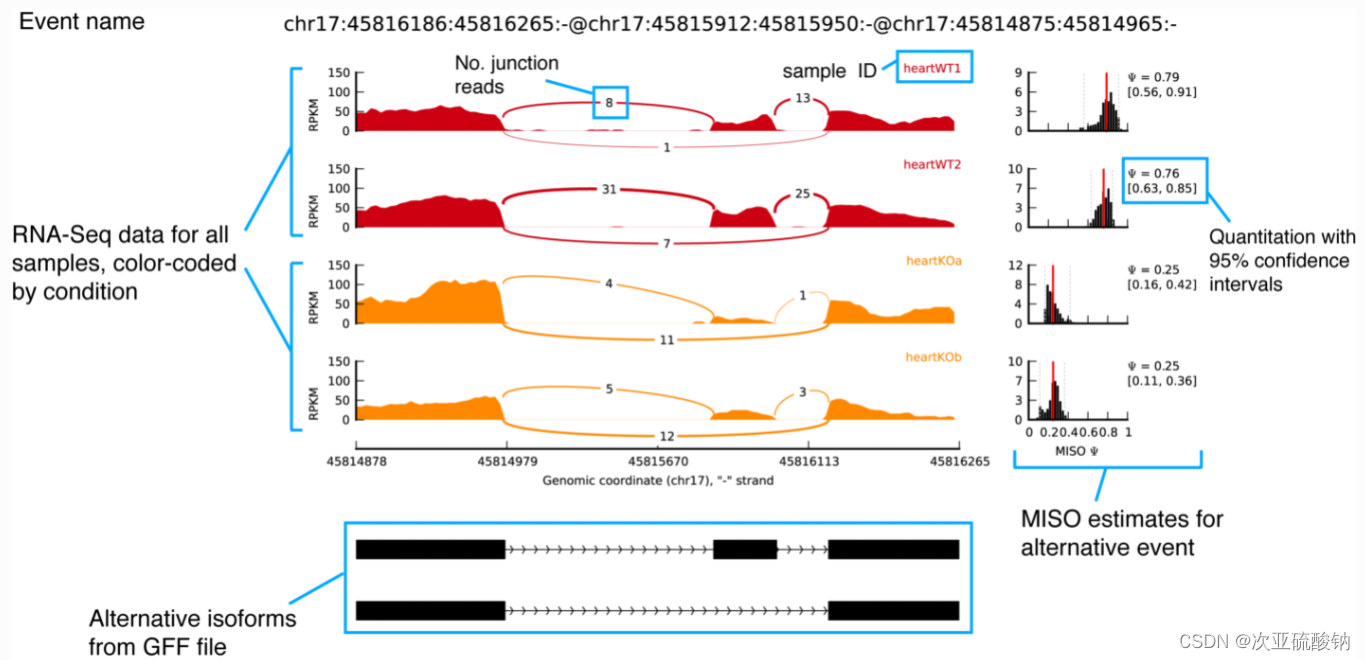
第一眼看到这图就被征服了!!太好看!这就是想要的效果!马上学起来!
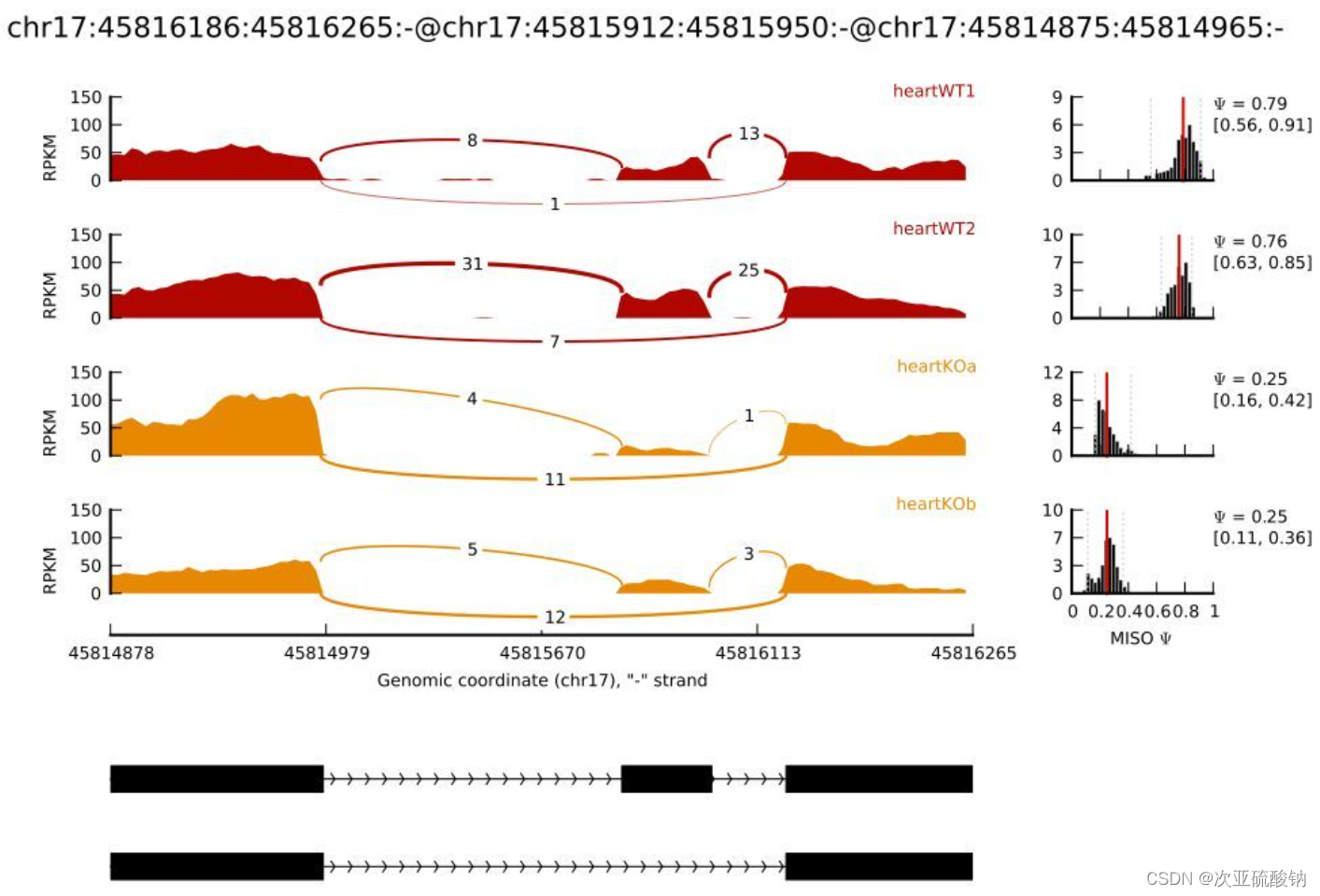
先解释下这种图,作者把这种图叫做sashimi_plot,因为长得和生鱼片比较像

下方的黑色矩形是从GFF中读取的外显子分布,左上方是每个样本中比对上exon的reads的可视化,采用了RPKM表示,不同剪切方式用曲线链接,曲线上标记的是比对上该区域的reads数目,不同分组的样本用不同颜色表示。右侧的图片是样本中对应的可变剪切的表达量值。
1.安装
miso需要依赖python和一些python的包,python建议2.7.x版本,我用的是2.7.15,直接用conda安装,miso和其他包就用pip安装:
conda install python=2.7.15
pip install misopy
pip install numpy
pip install scipy
pip install pysam
pip install matplotlib
2.构建索引
用自带的index_gff方法对GFF文件构建索引
index_gff --index ./final_gene.gff ./index_db
#index_db为输出文件夹
!需要注意BAM文件里的染色体号和GFF里必须对应,可能会出现一处是”chr3“,另一处是”3“的情况
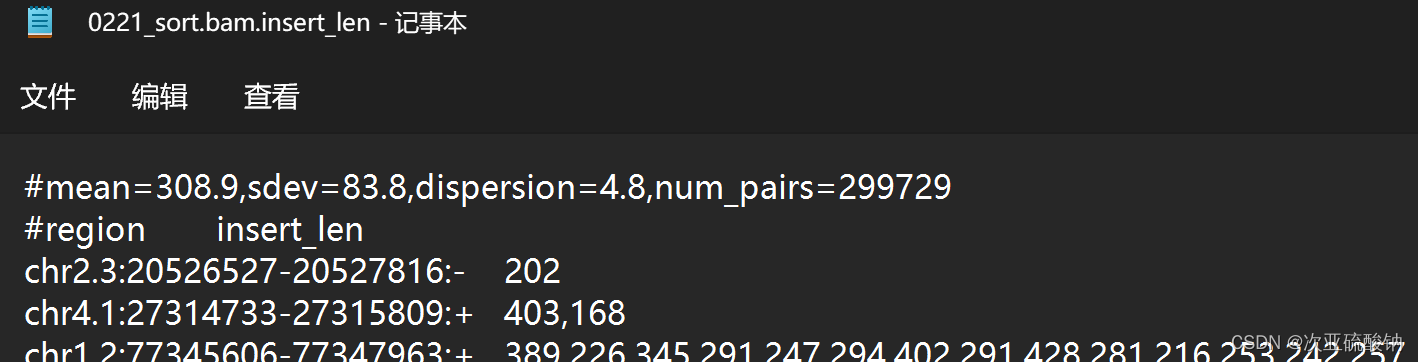
3.计算插入长度分布及其统计信息
通过exon_utils将reads对准长度的构成外显子,然后测量每对的插入长度来计算样品的插入长度分布。通过这种方式获得的插入长度集形成了一种分布,并且用pe_utils统计这种分布的数据
exon_utils --get-const-exons ./final_gene.gff \
--min-exon-size 1000 \
--output-dir ./exons/
exon_untils会在目标文件夹里输出一个至少1000个碱基长度的外显子GFF文件。pe_utils使用输出的GFF对BAM文件计算插入长度
pe_utils --compute-insert-len ./0221_sort.bam \
./exons/final_gene.min_1000.const_exons.gff \
--output-dir ./insert-dist/
在输出的.insert_len文件中首行可以看到插入长度分布的均值、标准差、离散度以及用于估计分布和计算这些值的读对数。均值和标准差在后面运行时有用。
4.运行
运行miso需要一个设置文件,官方自带了一个settings/miso_settings.txt,直接用也行,一般参数默认就ok,具体各个参数的作用看官网吧
[data]
filter_results = True
min_event_reads = 20
strand = fr-unstranded
[cluster]
cluster_command = qsub
[sampler]
burn_in = 500
lag = 10
num_iters = 5000
num_processors = 4
官网有单端测序的示例,我用的是双端测序。BAM文件要排序过的,并且和bai索引要处在同一个文件夹下,这里因为我是提交集群运算,去掉了num_processors = 4,用的自己的setting文件。read-len是读长,这个数值可以从质控的结果里看到。paired-end后面的两个数字分别是插入片段长度的均值和方差。
miso --run ./index_db ./0221_sorted.bam \
--output-dir ./out_dir \
--read-len 150 \
--paired-end 308.9 83.8 \
--settings-filename ./miso_setting.txt
到这步可以说成功一半了!然后接下来…
就没有然后了😭
因为miso不支持混合读长,报错了
MISO does not support mixed read lengths. Please trim your reads to a uniform length and re-run or run MISO separately on each read length. Proceeding anyway, though this may result in errors or inability to match reads to your annotation.
Read lengths were: 128,129,130,131,132,133,134,135,136,137,138,139,140,141,142,143,144,145,146,147,148,149,150,55,56,57,63,67,70,71,72,73,75,76,77,78,79,80,81,82,83,84,85,86,87,88,89,90,91,92,93,94,95,96,97,98,99,100,101,102,103,104,105,106,107,108,109,110,111,112,113,114,115,116,117,118,119,120,121,122,123,124,125,126,127
搜了一下,目前这个问题有两种解决办法:
1.修剪读长,修改FATSQ或BAM文件,全部改到一样的长度
2.等更新,等它更新到能支持混合读长…
参考:
https://miso.readthedocs.io/en/fastmiso/#installation
http://blog.sina.com.cn/s/blog_83f77c940102y19m.html
https://www.jianshu.com/p/18dd5396bb77
https://cloud.tencent.com/developer/article/1625312
https://www.biostars.org/p/175838/
https://miso-users.mit.narkive.com/Xh5rX1Cz/install-miso-using-pip-and-mixed-read-length-bams
https://github.com/yarden/MISO/issues/94
rMATS
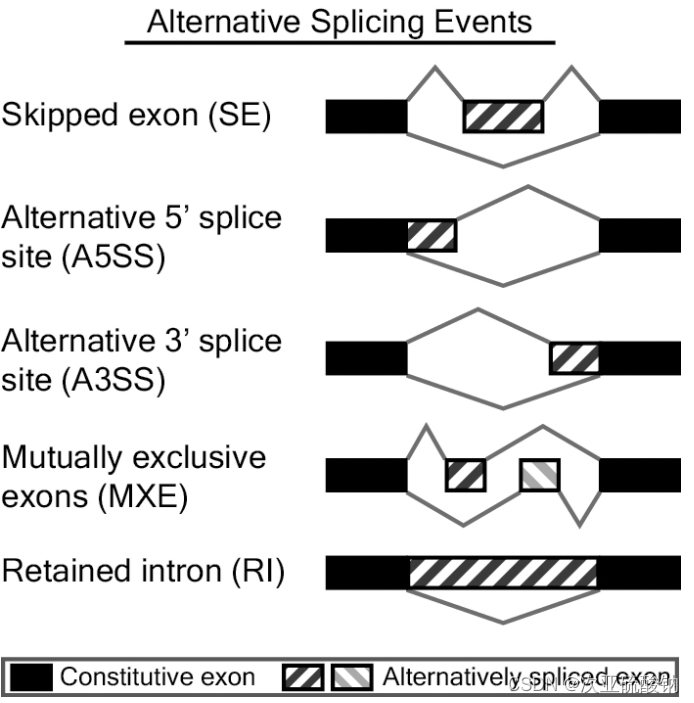
miso行不通就换一个效果差不多的。rmats可以识别5种可变剪接事件:Skippedexon (SE) 外显子跳跃、Alternative5’ splice site (A5SS) 5’端可变剪切、Alternative3’ splice site (A3SS) 3’端可变剪切、Mutuallyexclusive exons (MXE) 互斥可变外显子、Retainedintron (RI) 内含子保留
1.安装
因为要用到专门可视化rmats的rmats2sashimiplot,所以就先一起装了,先解决两个软件的环境。
conda install python
conda install samtools
conda install bedtools
#python安装成功后,用pip安装
pip install numpy
pip install scipy
pip install matplotlib
pip install pysam
再用conda安装rmats,安装成功后用rmats.py -h命令,看看能不能输出帮助文档
conda install rmats
rmats2sashimiplot需要去官网下载、解压,复制到服务器。
进入到rmats2sashimiplot-master的文件夹里,用命令chmod 777 setup.py开放全部权限。执行python ./setup.py install安装。输入rmats2sashimiplot -h能出现帮助文档即成功
如果运行rmats时出现缺
libgfortran.so.3或libgsl.so.0的报错,请看这里https://www.jianshu.com/p/451b7e84bbe5
2.运行
rmats支持FASTQ和BAM文件输入,用FASTQ输入的话,需要再安装STAR,好像FASTQ输入不保存中间的BAM文件,建议还是用BAM输入。我是用Hitsat2构建好的BAM作为输入,BAM文件要排序过的,最好提前构建索引,并且放在同一个目录下。
rmats也是要求BAM文件和GTF染色体号要对应,我的数据出乎意料的一致,但也找到了处理方法
sed -i '2,$s/\tchr/\t/g' *MATS*txt
#-i表示直接修改文件内容,2,$表示从第二行到最后一行(第一行是列名,不能替换),g表示全局
接下来把BAM文件的文件名写入b1.txt和b2.txt,一个实验组,另一个对照组。注意各个bam文件之间用逗号隔开,但不能放空格,用英文逗号,建议用绝对路径。b1和b2文件需要转为Linux格式,rmats局限在只能两个分组,多个分组比对需要多运行几次.
echo ./1.bam,./2.bam > b1.txt
echo ./3.bam,./4.bam > b2.txt
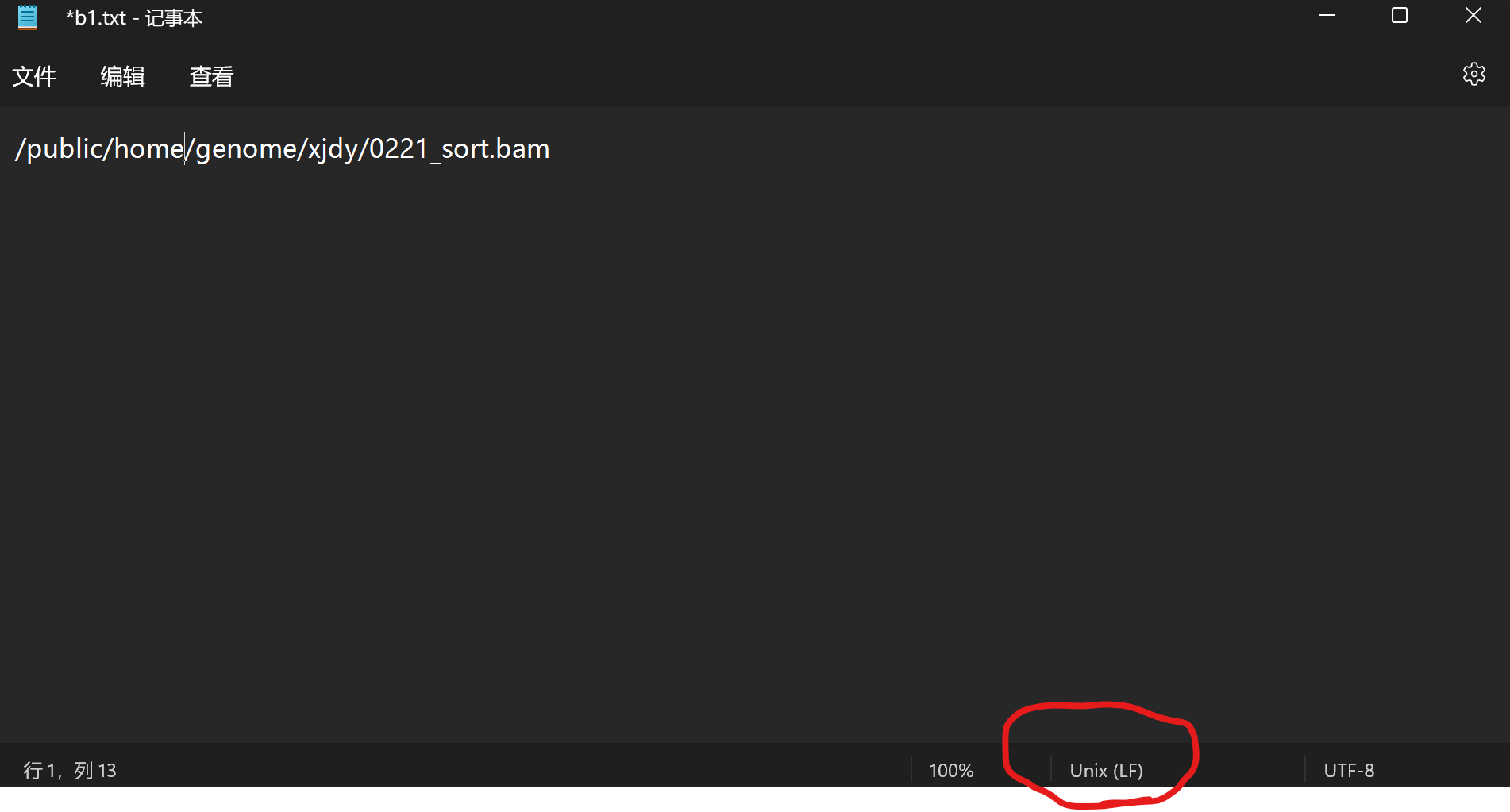
运行时的--b1、--b2也建议用绝对路径输入。--grf是参考基因组注释。--od是存放输出结果的文件夹。-t表示双端测序。--nthread线程数。--readLength测序的读长,可以从质控报告里看到。--tmp缓存文件存储目录。
用
gffread *.gff -T -o *.gtf可以转换gff文件
rmats.py \
--b1 ./b1.txt \
--b2 ./b2.txt \
--gtf ./final_gene.gtf \
--od output_rmats \
-t paired \
--nthread 4 \
--readLength 150 \
--tmp out_rmats
从日志文件中可以看比对到的类型:

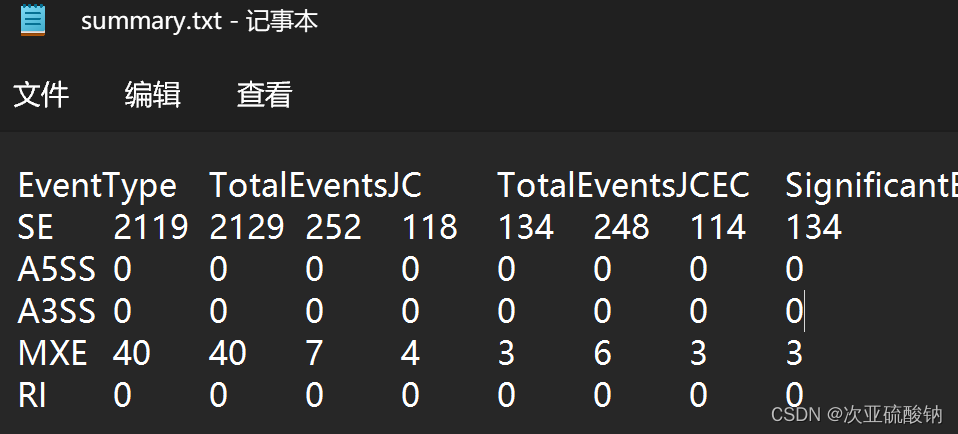
输出文件里的output_rmats/summary.txt里也有个比对的类型,但是没搞清楚为什么两个文件内数量对不上,大致是一样的,看个趋势没问题
3.结果解读
/output_rmats/里会输出这几种文件:[AS_Event].MATS.JC.txt 最常用的结果文件,统计了跨剪切位点的reads,AS_Event包括A3SS、A5SS、MXE、RI、SE。fromGTF.[AS_Event].txt从GTF和RNA衍生出的所有已鉴定的替代剪接(AS)事件。fromGTF.novelJunction.[AS_Event].txt选择性剪接 (AS) 事件,仅在考虑 RNA 后才被识别(而不是单独分析 GTF),这不包括具有未注释剪接位点的事件。fromGTF.novelSpliceSite.[AS_Event].txt该文件仅包含那些包含未注释拼接位点的事件。仅在启用–novelSS参数时相关。JC.raw.input.[AS_Event].txt仅包括跨rmat定义的交汇点的读取的事件计数(交汇点计数)。summary.txt所有AS事件类型的简要摘要。包括总事件计数和重要事件计数。默认情况下,如果FDR<=0.05,则将事件视为重要事件。通过运行rMATS_P/asumy.py,可以使用不同的标准重新生成摘要。tmp包含由准备步骤生成的中间文件。
JC文件会有一个对应的JCEC文件,二者的区别在于JC考虑跨越剪切位点的reads,而JCEC不仅考虑前者的reads还考虑到只比对到Fig 1中条纹的区域(也就是说没有跨越剪切位点的reads),但是我们一般使用JC.raw.input.AS_Event.txt的结果就够了(如果只是单纯的比较两组样品间可变剪切的差异的话)
以MXE.MATS.JC.txt为例说明每列的意义:
ID GeneID geneSymbol chr strand 1stExonStart_0base 1stExonEnd 2ndExonStart_0base 2ndExonEnd upstreamES upstreamEE downstreamES downstreamEE ID IJC_SAMPLE_1 SJC_SAMPLE_1 IJC_SAMPLE_2 SJC_SAMPLE_2 IncFormLen SkipFormLen PValue FDR IncLevel1 IncLevel2 IncLevelDifference
0 "MS.gene23798" NA chr8.4 - 30758609 30758704 30759122 30759182 30758025 30758095 30760455 30760527 0 1 11 7 9 209 244 0.0120878457309 0.0604392286545 0.096 0.476 -0.38
1 "MS.gene61989" NA chr7.2 - 80697619 80697769 80704270 80704420 80697113 80697232 80705567 80706851 1 1 1 0 3 298 298 0.102057409464 0.34019136488 0.5 0.0 0.5
- ID: 官网描述“rMATS event id”,其实就是序号
- GenelD: 可变剪接事件所在基因编号
- geneSymbol: 可变剪接事件所在基因名称
- chr: 可变剪接事件所在染色体
- strand: 可变剪接事件所在染色体链的方向
- 1stExonStart_0base: 第一个可变剪接事件跳跃外显子的起始位置,以0开始计数
- 1stExonEnd: 第一个可变剪接事件跳跃外显子的终止位置
- 2ndExonStart_0base:第二个可变剪接事件跳跃外显子的起始位置,以0开始计数
- 2ndExonEnd: 第二个可变剪接事件跳跃外显子的终止位置
- upstreamES: 可变剪接事件跳跃外显子的上游exon起始位置
- upstreamEE: 可变剪接事件跳跃外显子的上游exon终止位置
- downstreamES: 可变剪接事件跳跃外显子的下游exon起始位置
- downstreamEE: 可变剪接事件跳跃外显子的下游exon终止位置

- ID: 同上
- IJC_SAMPLE_1: 样本一在inclusion junction(IJC)下的count数,重复样本的结果以逗号分隔
- SJC_SAMPLE_1: 样本一在skipping junction(SJC)下的count数,重复样本的结果以逗号分隔
- IJC_SAMPLE_2: 样本二在inclusion junction(IJC)下的count数,重复样本的结果以逗号分隔
- SJC_SAMPLE_2: 样本二在skipping junction(SJC)下的count数,重复样本的结果以逗号分隔
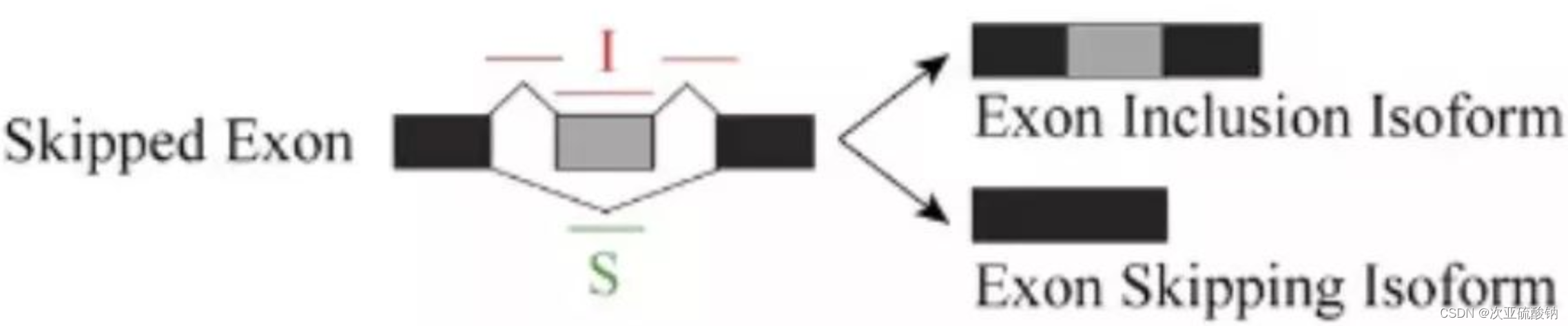
- IncFormLen: 可变剪接事件Exon Inclusion Isoform的有效长度
- SkipFormLen: 可变剪接事件Exon Skipping Isoform的有效长度
- PValue: 两组样本间可变剪接事件表达差异显著性p值
- FDR: 可变剪接事件表达差异显著性FDR值
- IncLevel1: 处理组可变剪接事件Exon Inclusion Isoform在两个Isoform总表达量的比值
- IncLevel2: 对照组可变剪接事件Exon Inclusion Isoform在两个Isoform总表达量的比值
- IncLevelDifference: IncLevel1与IncLevel2的差值
4.可视化
rmats2sashimiplot 支持SAM或BAM作为输入,一般用BAM
有三种输出方法,先介绍常用的参数:--b1和--b2是经过排序且有索引的;-t是rmats输出的可变剪接类型,有五个选项SE RI A3SS A5SS MXE;-c:染色体的座标 (染色体-正负链-开始-终止 + gff3格式的注释文件);-e是对应-t所选参数的文件。这里的需要可视化SE.MATS.JC.txt的文档,看到有教程说“ 里面有几条内容,就会生成几个图片,所以需要自己去挑选一下,挑选的时候,尽量不要选存在 NA 的值 ”,实际使用的时候数量不一致,可能是存在NA?--l1、--l2输出对照组和实验组对应的名字(可以自己起);-o结果输出的文件夹;--intron_s指定图片中intron的比例
-t、-e的方法不能和-c连用,亲测同时用只会按前者方式输出
有个教程提供了循环的方法:https://www.jianshu.com/p/451b7e84bbe5
1)文件输入
这种方法把rmats输出的某个可变剪接事件作为输入,会可视化事件里的全部基因,适合看整体的差异变化。一次只能选一种文件
rmats2sashimiplot --b1 ./0221_sort.bam \
--b2 ./0225_sort.bam \
-t SE \
-e ./output_rmats/SE.MATS.JC.txt \
--l1 0221_sort \
--l2 0225_sort \
-o ./plot

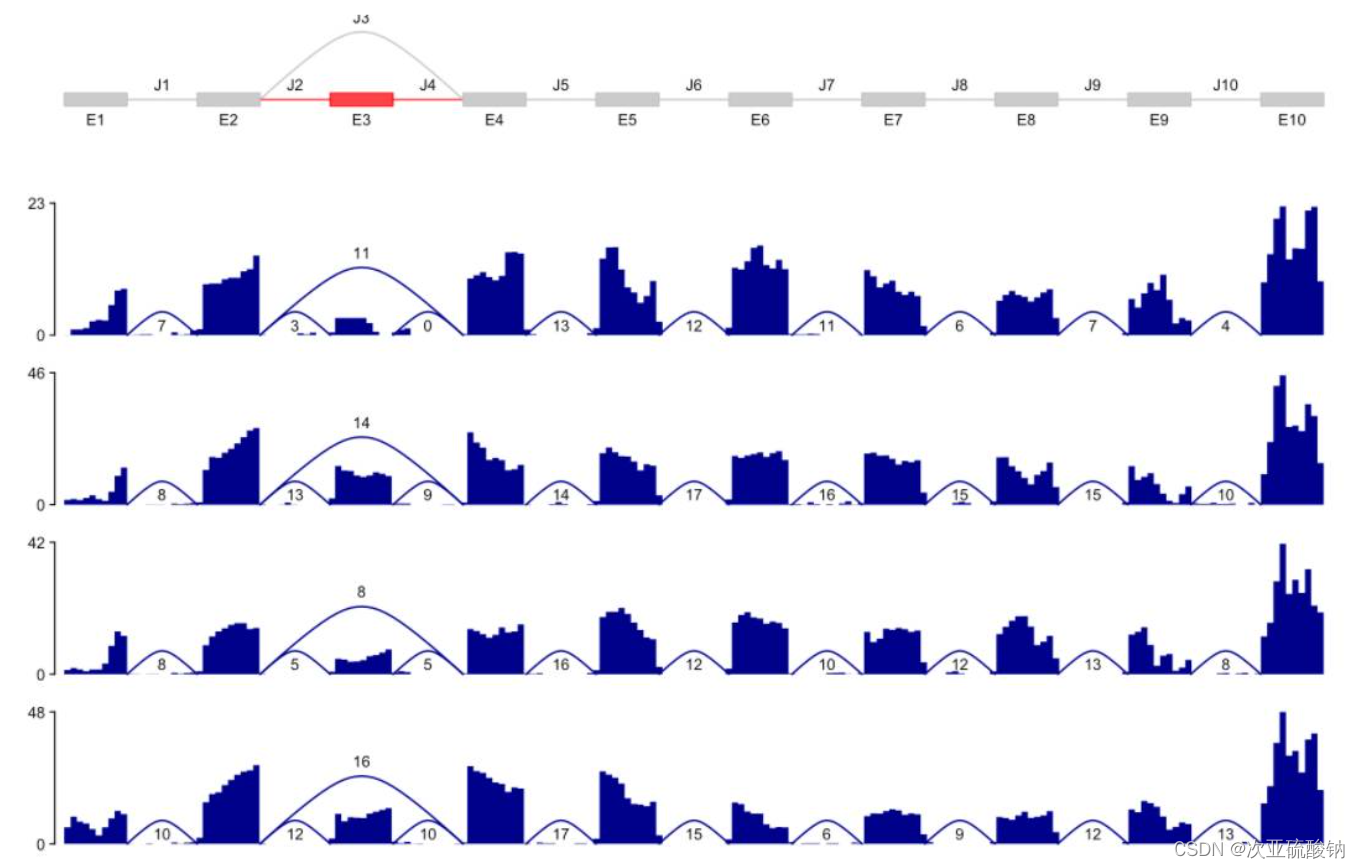
可以看到没有miso右侧的柱状图,这是因为rmats中只会计算可变剪切事件的inclusion level值,并没有提供95%执行区间的值。图中的IncLevel值是直接从rmats的输出结果中读取的,所以二者是一致的。
对于曲线上方标记的reads数目,和rmats输出结果中的IJC和SJC是不同的,因为是两个软件统计的结果,从配置文件可以看出,只提供了bam文件给miso,所以图上的reads数是由miso这个软件计算得到的,自然和rmats计算的reads数目有所出入,但是只是微小差异,总体的趋势还是一致的。
对于rmats2sashimiplot产生的图片,我们不需要太过于纠结reads数目和rmats对应不上,只需要看reads分布的大体趋势即可,重点是关注不同样本中IncLevel的差异。
2)基因坐标输入
使用-c以基因坐标的形式输入,结尾坐标会有时-1有时+1,输出文件只有一个。
rmats2sashimiplot --b1 ./0221_sort.bam \
--b2 ./0225_sort.bam \
-c chr8.4:-:30755930:30760752:./final_gene.gff \
--l1 0221_sort \
--l2 0225_sort \
-o ./plot
建议还是从输出的可变剪接事件中找到目标基因,再用这种方式可视化。试过从注释文件里随便找个基因坐标,输出失败,应该是要有可变剪接才能可视化。这里随便选了一段30758609:30760527结果没有东西输出

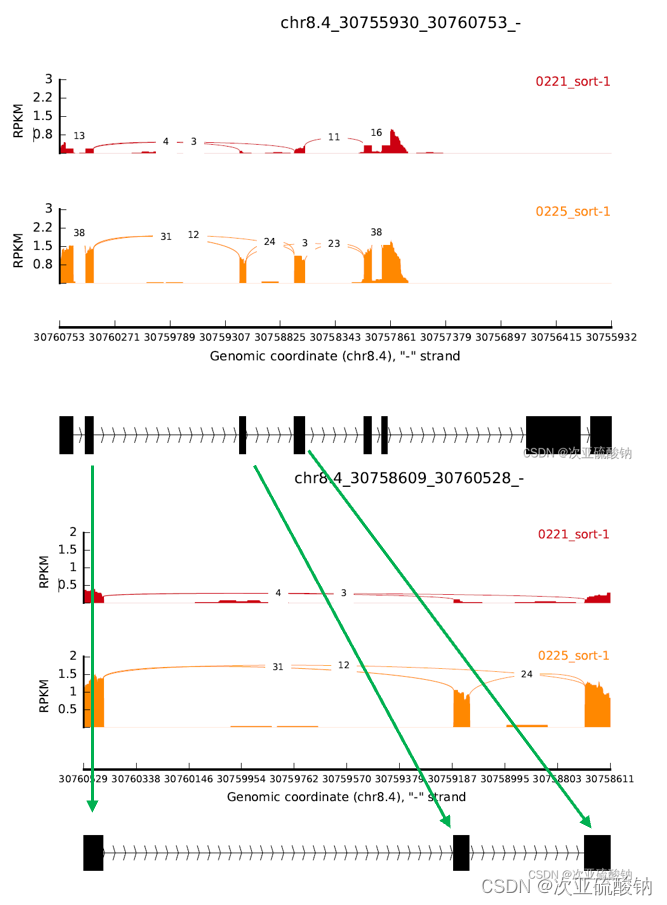
这个坐标是从GFF文件中取出的完整基因坐标,所以会有大段的空,猜测是UTR区

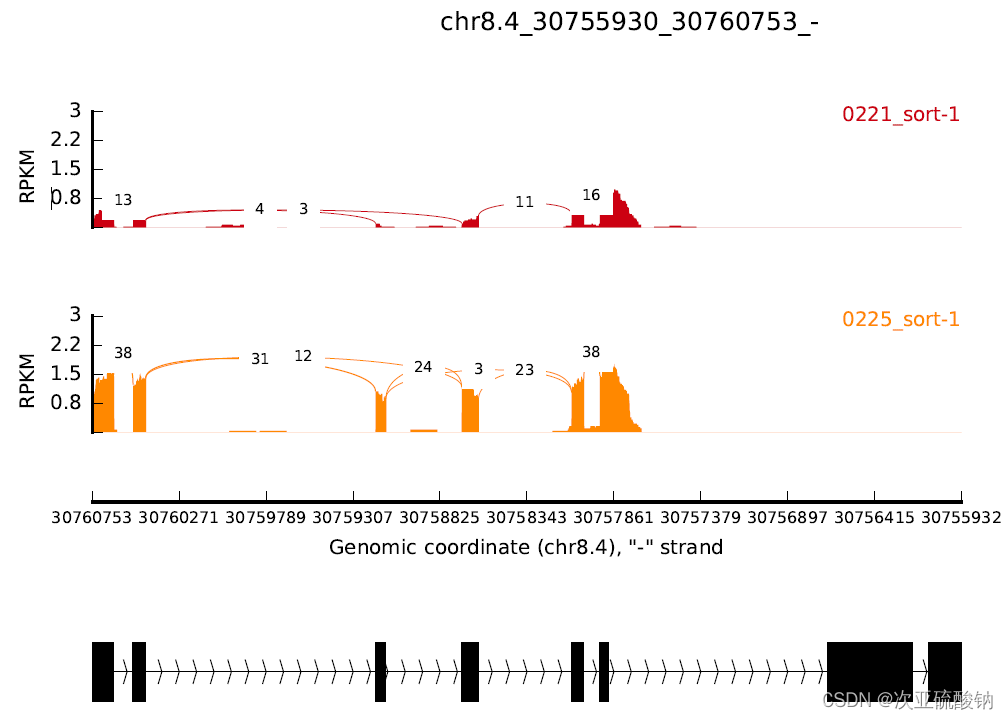
如果用可变剪接事件里的坐标就会填满整个图,不会留空。这里用的是1stExonStart_0base和downstreamEE
ID GeneID geneSymbol chr strand 1stExonStart_0base 1stExonEnd 2ndExonStart_0base 2ndExonEnd upstreamES upstreamEE downstreamES downstreamEE ID IJC_SAMPLE_1 SJC_SAMPLE_1 IJC_SAMPLE_2 SJC_SAMPLE_2 IncFormLen SkipFormLen PValue FDR IncLevel1 IncLevel2 IncLevelDifference
0 "MS.gene23798" NA chr8.4 - 30758609 30758704 30759122 30759182 30758025 30758095 30760455 30760527 0 1 11 7 9 209 244 0.0120878457309 0.0604392286545 0.096 0.476 -0.38
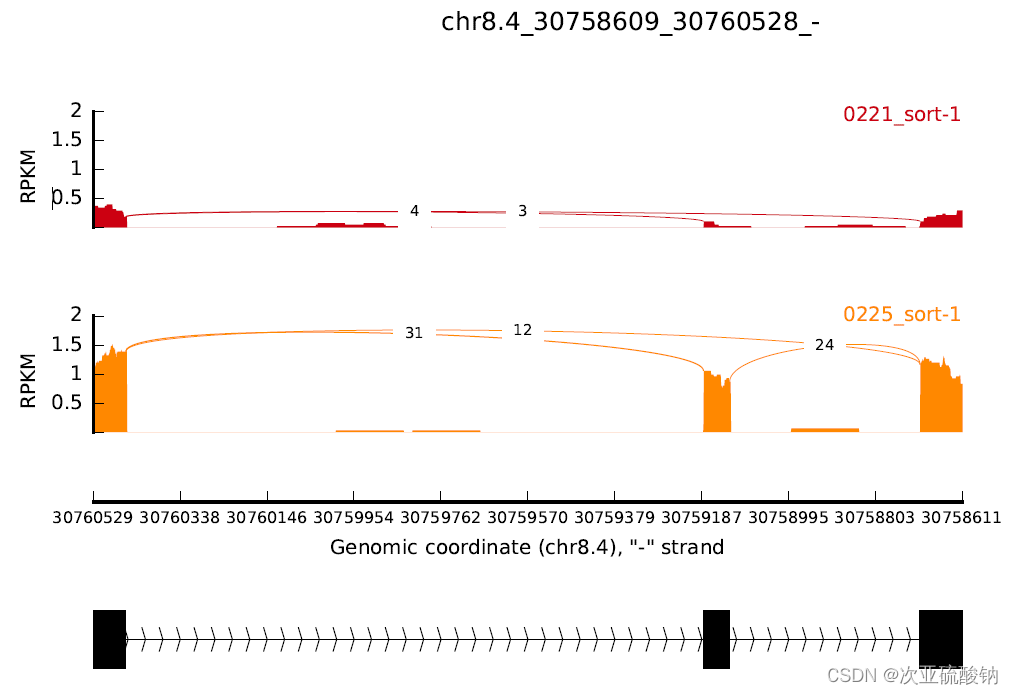
其实也是能看出两种不同输出的对应关系,GFF中完整基因坐标的范围会更大
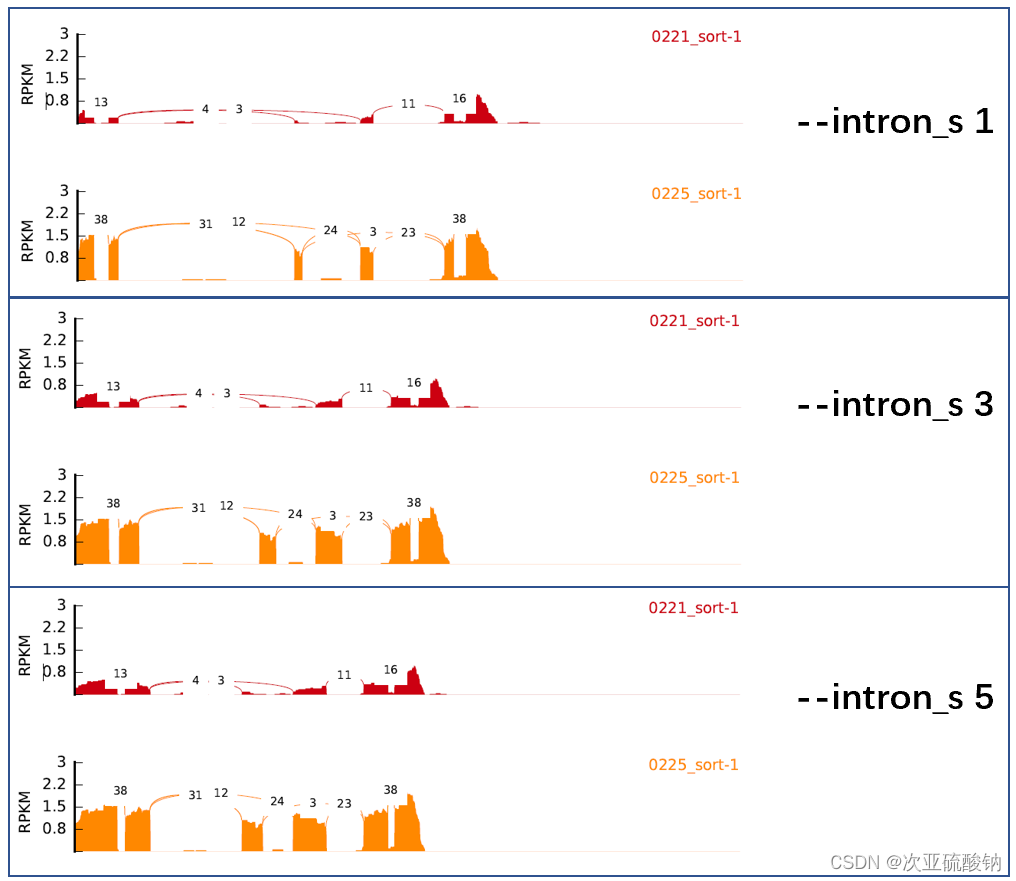
最后说下--intron_s的用法,这个能够调整显示峰值的比例,默认是1,下图是用同一个基因坐标测试不同数值的比例效果
3)样本组输入
当一个样品有多个重复的时候,可以用--group-info对它们进行合并,计算均值,作为一个整体分析。这部分还没实操过,用的官网例子
rmats2sashimiplot \
--b1 ./sample_1_replicate_1.bam,./sample_1_replicate_2.bam,./sample_1_replicate_3.bam \
--b2 ./sample_2_replicate_1.bam,./sample_2_replicate_2.bam,./sample_2_replicate_3.bam \
-t SE \
-e ./SE.MATS.JC.txt \
--l1 SampleOne \
--l2 SampleTwo \
--exon_s 1 \
--intron_s 5 \
-o test_grouped_output \
--group-info grouping.gf
###------------------------
cat grouping.gf
group1name: 1-2
group2name: 3-6
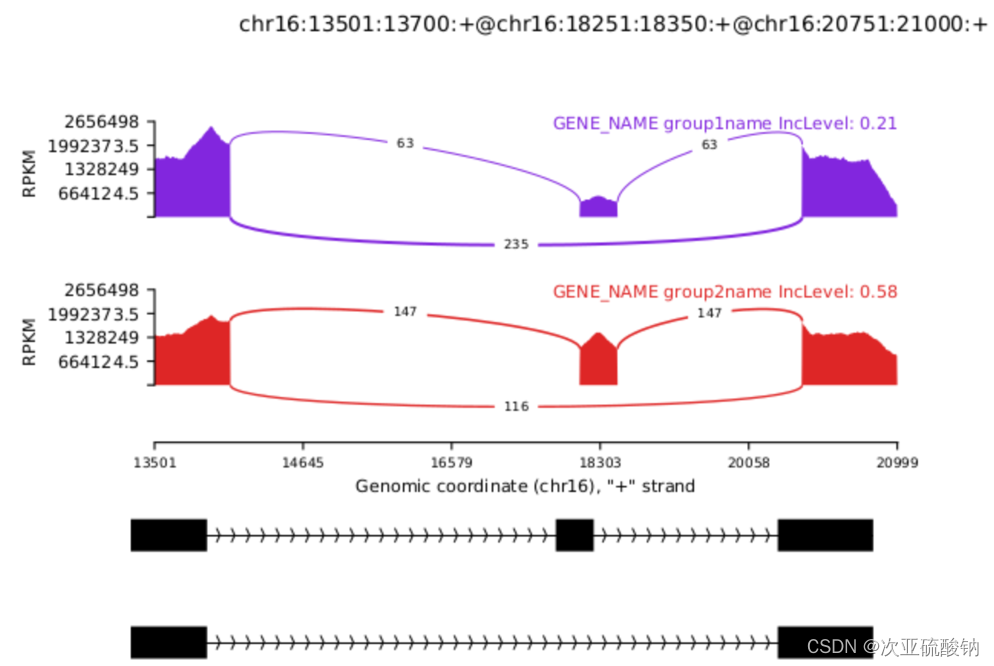
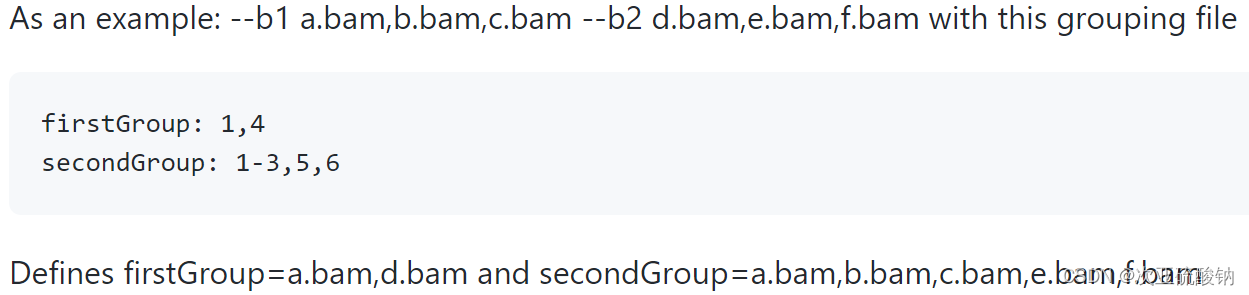
--b支持bam和sam作为输入。这里的*.gf文件是用来区别不同分组,用echo写入分组信息,有两种表示方法,,单独分隔和-按范围取。举个栗子:如果有6个输入--b1 a.bam,b.bam,c.bam --b2 d.bam,e.bam,f.bam这里abc是实验组的3个重复,def是对照组的3个重复,就可以用
test: 1,2,3
control: 4,5,6
这里test和control可以随意命名,会显示在图上。也可以用-来表示一样的效果
test: 1-3
control: 4-6
需要注意输入文件的顺序非常重要
参考:
https://blog.csdn.net/weixin_51192038/article/details/122135310
https://blog.csdn.net/weixin_51192038/article/details/116131250
https://www.jianshu.com/p/451b7e84bbe5
https://www.i4k.xyz/article/weixin_51192038/116131250
https://www.jianshu.com/p/abab2f188172
https://blog.csdn.net/weixin_40007804/article/details/112145840
https://cloud.tencent.com/developer/article/1587437?from=article.detail.1625308
https://cloud.tencent.com/developer/article/1625313?from=article.detail.1625308
https://zpliu.gitbook.io/booknote/ke-bian-jian-qie/di-wu-ci-fen-xi/hui-zhi-ke-bian-jian-qie
http://rnaseq-mats.sourceforge.net/index.html
https://github.com/Xinglab/rmats-turbo/blob/v4.1.2/README.md#all-arguments
https://www.bioinfo-scrounger.com/archives/611/
https://zhuanlan.zhihu.com/p/228962609
https://github.com/Xinglab/rmats2sashimiplot#sam-files-with-rmats-event
彩蛋
rmats官网推荐了一些可以协同使用的软件:rMAPS: RNA Map Analysis and Plotting Server:用来分析rMATS的SE事件周围的RNA结合蛋白(RBPs)结合位点;rMATS-STAT:用于测试差异剪接的独立统计模型;PrimerSeq: Primer Seek in RNA-seq:用于设计rMATS输出的RT-PCR引物。有空再摸索一下这些~

















 904
904

 被折叠的 条评论
为什么被折叠?
被折叠的 条评论
为什么被折叠?
 到【灌水乐园】发言
到【灌水乐园】发言









