







 本文介绍了单细胞RNA测序数据的分析流程,包括准备R包和数据、构建CellDataSet对象、质控、聚类、差异分析和推断发育轨迹。使用scRNAseq的示例数据fluidigm,过滤低表达基因,筛选平均表达量>0.1的基因聚类,还可进行差异分析和发育轨迹推断。
本文介绍了单细胞RNA测序数据的分析流程,包括准备R包和数据、构建CellDataSet对象、质控、聚类、差异分析和推断发育轨迹。使用scRNAseq的示例数据fluidigm,过滤低表达基因,筛选平均表达量>0.1的基因聚类,还可进行差异分析和发育轨迹推断。
0.准备R包和数据
library(monocle)
library(scRNAseq)
library(dplyr)
使用scRNAseq里面的示例数据fluidigm。这个示例数据本身是一个对象,我们又需要从中提取出来自己想要的组成部分,然后组成新的对象。
曾经fluidigm是一个可以用data提取的数据,后来包更新了,只能用下面的函数来调用咯
fluidigm = ReprocessedFluidigmData()
# Set assay to RSEM estimated counts
assay(fluidigm) <- assays(fluidigm)$rsem_counts
ct <- floor(assays(fluidigm)$rsem_counts)
ct[1:4,1:4]
## SRR1275356 SRR1274090 SRR1275251 SRR1275287
## A1BG 0 0 0 0
## A1BG-AS1 0 0 0 0
## A1CF 0 0 0 0
## A2M 0 0 0 33
dim(ct) #count矩阵,行是基因,列是细胞
## [1] 26255 130
可以看到这里是有130个细胞,两万六千多个基因。
sample_ann <- as.data.frame(colData(fluidigm))
table(sample_ann$Biological_Condition)
##
## GW16 GW21 GW21+3 NPC
## 52 16 32 30
细胞注释数据里提供了Biological Condition,后续的分析是以它为中心来分析的。
1.构建CellDataSet对象
gene_ann <- data.frame(
gene_short_name = row.names(ct),
row.names = row.names(ct)
)
pd <- new("AnnotatedDataFrame",
data=sample_ann)
fd <- new("AnnotatedDataFrame",
data=gene_ann)
cds <- newCellDataSet(
ct,
phenoData = pd,
featureData =fd,
expressionFamily = negbinomial.size(),
lowerDetectionLimit=1)
class(cds)
## [1] "CellDataSet"
## attr(,"package")
## [1] "monocle"
## 为了电脑的健康,我这里选择小数据集。
cds <- estimateSizeFactors(cds)
cds <- estimateDispersions(cds)
2.质控
过滤前是26255 features, 130 samples,筛掉在5个以下细胞中表达的基因。这个标准是非常低的咯。
dim(cds)
## Features Samples
## 26255 130
cds <- detectGenes(cds, min_expr = 0.1)
head(fData(cds))
## gene_short_name num_cells_expressed
## A1BG A1BG 10
## A1BG-AS1 A1BG-AS1 2
## A1CF A1CF 1
## A2M A2M 21
## A2M-AS1 A2M-AS1 3
## A2ML1 A2ML1 9
k = fData(cds)$num_cells_expressed>=5;table(k)
## k
## FALSE TRUE
## 12870 13385
cds <- cds[k,]
cds
## CellDataSet (storageMode: environment)
## assayData: 13385 features, 130 samples
## element names: exprs
## protocolData: none
## phenoData
## sampleNames: SRR1275356 SRR1274090 ... SRR1275261 (130 total)
## varLabels: NREADS NALIGNED ... num_genes_expressed (30 total)
## varMetadata: labelDescription
## featureData
## featureNames: A1BG A2M ... ZZZ3 (13385 total)
## fvarLabels: gene_short_name num_cells_expressed
## fvarMetadata: labelDescription
## experimentData: use 'experimentData(object)'
## Annotation:
dim(cds)
## Features Samples
## 13385 130
过滤基因后是:13385 features, 130 samples,很低的标准也过滤掉了一半的基因。
fivenum(pData(cds)$NREADS)
## [1] 91616 232899 892209 8130850 14477100
fivenum(pData(cds)$num_genes_expressed)
## [1] 1418.0 2961.0 3841.5 5381.0 8221.0
NREADS展示了每个细胞中总共多少条reads,num_genes_expressed展示了每个细胞中有多少个基因表达量不为0。可以根据这两个指标设置阈值去过滤细胞。在这里不需要过滤。
3.聚类
筛选平均表达量 > 0.1的基因用于聚类
disp_table <- dispersionTable(cds)
unsup_clustering_genes <- subset(disp_table, mean_expression >= 0.1)
cds <- setOrderingFilter(cds, unsup_clustering_genes$gene_id)
碎石图,降维和细胞聚类
plot_pc_variance_explained(cds,
return_all = F,
max_components = 20)
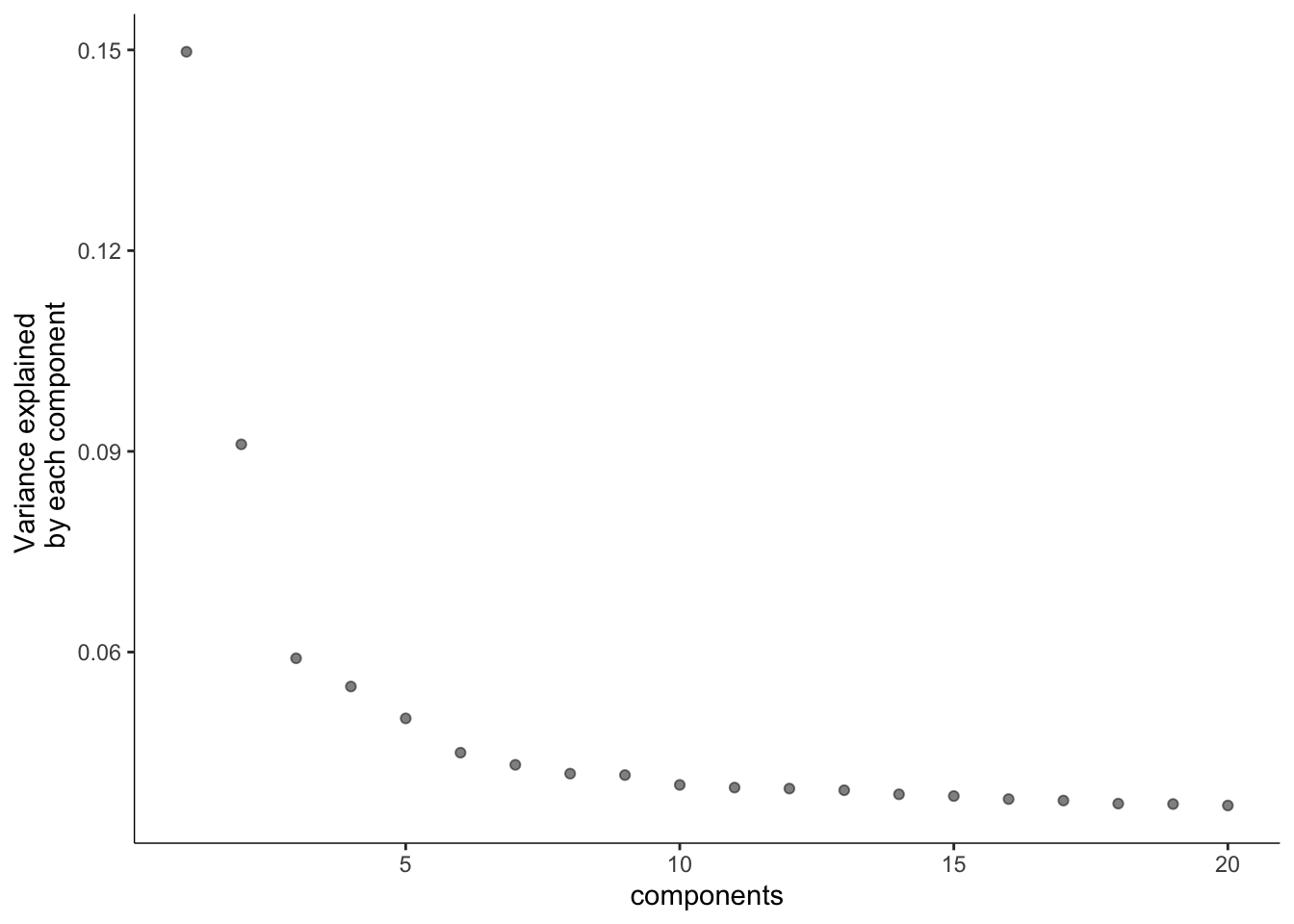
# 其中 num_dim 参数选择基于上面的PCA图
cds <- reduceDimension(cds,
max_components = 2,
num_dim = 6,
reduction_method = 'tSNE', verbose = T)
cds <- clusterCells(cds, num_clusters = 4)
## Distance cutoff calculated to 0.5395435
plot_cell_clusters(cds, 1, 2,
color = "Biological_Condition")
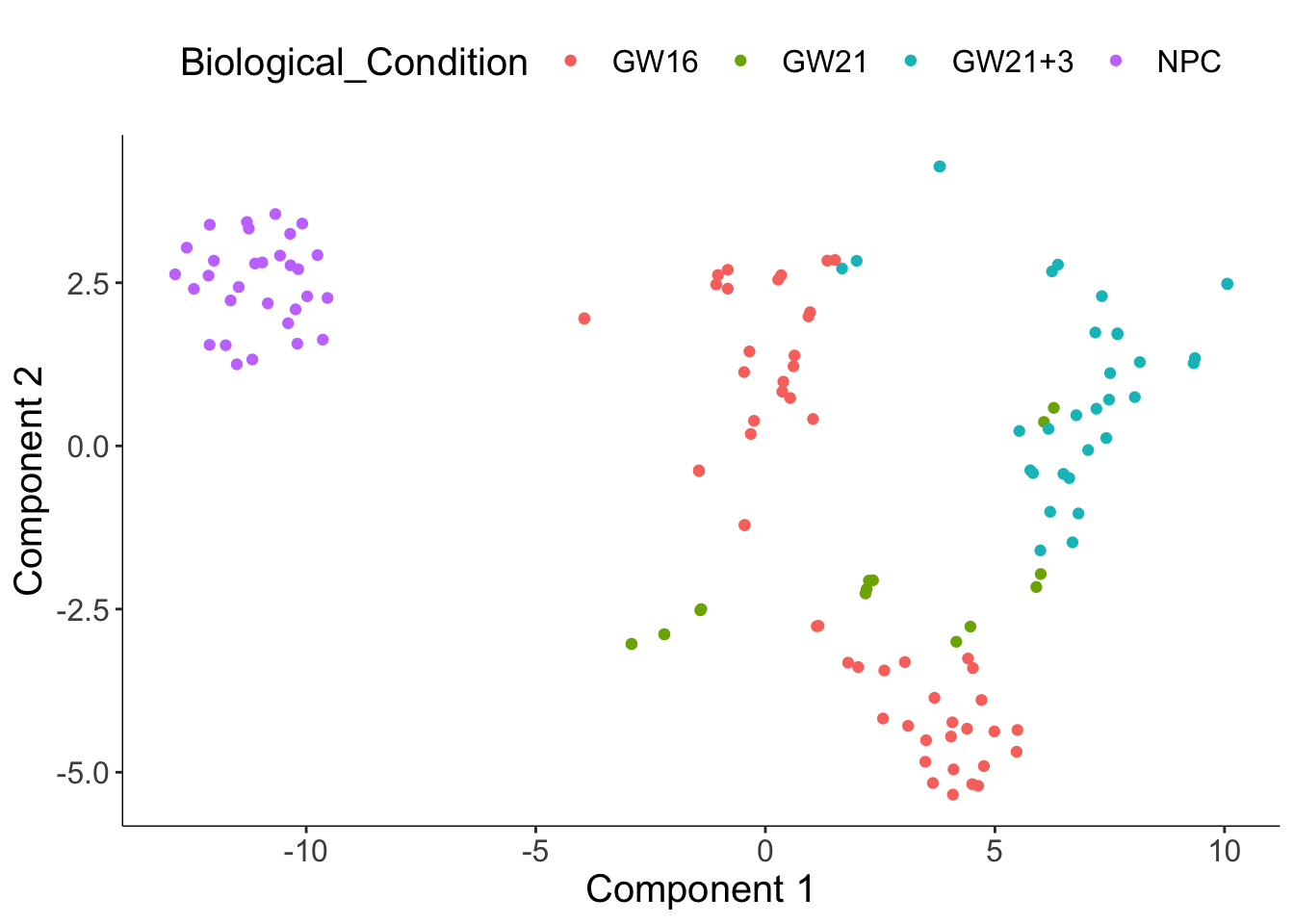
发现聚类结果与Biological Condition高度吻合。如果不指定颜色的话,是按照聚出来的细胞簇来分配颜色。
如果有需要去除的干扰因素,可以使用reduceDimension里的 residualModelFormulaStr参数,去除干扰因素再聚类。
4.差异分析
找差异基因的过程给做成函数咯。
diff_test_res <- differentialGeneTest(cds,
fullModelFormulaStr = "~Biological_Condition")
# Select genes that are significant at an FDR < 10%
sig_genes <- subset(diff_test_res, qval < 0.1)
dim(sig_genes)
## [1] 6427 7
head(sig_genes[,c("gene_short_name", "pval", "qval")] )
## gene_short_name pval qval
## A1BG A1BG 6.759792e-04 2.362397e-03
## A2M A2M 5.659195e-08 5.852474e-07
## AACS AACS 3.329596e-03 9.677356e-03
## AADAT AADAT 1.403810e-02 3.422585e-02
## AAGAB AAGAB 1.717061e-07 1.540937e-06
## AAMP AAMP 8.820175e-05 3.849301e-04
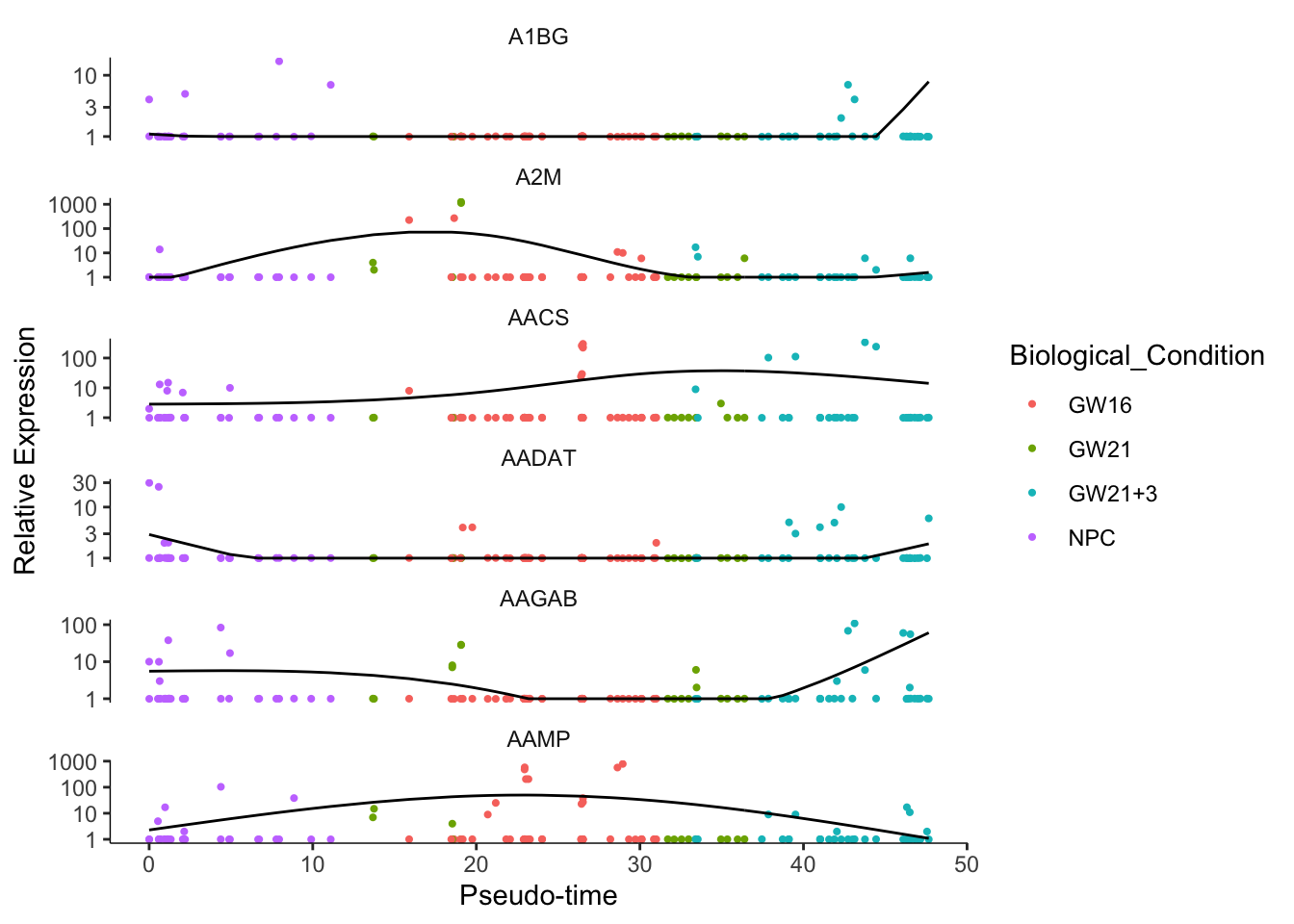
还可以很方便的指定基因画表达量散点图。横坐标没有意义,每个点是一个细胞。
cg = head(sig_genes$gene_short_name)
plot_genes_jitter(cds[cg,],
grouping = "Biological_Condition",
ncol= 2)
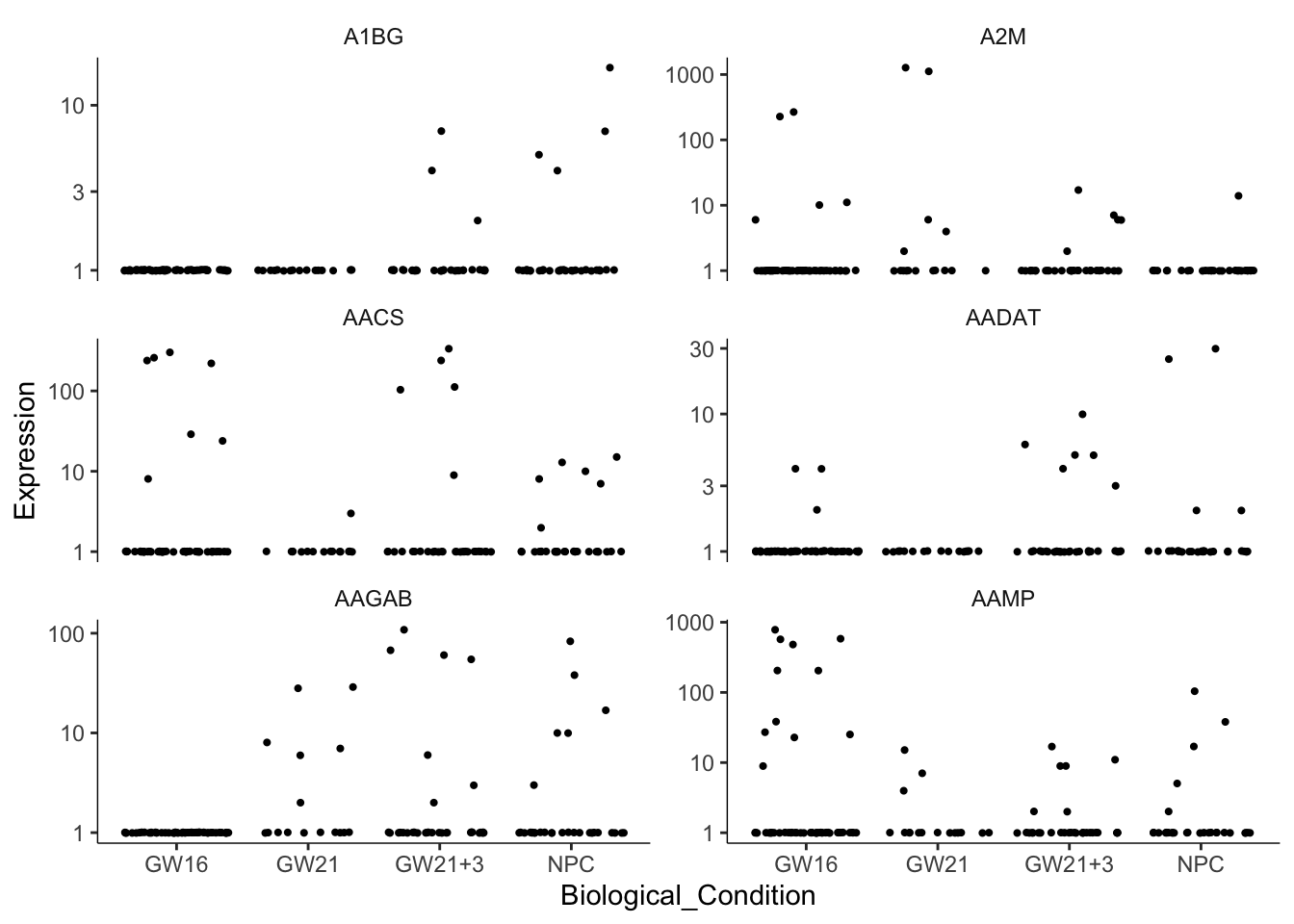
5.推断发育轨迹
挑选差异显著的基因(q<0.01)做降维,给细胞排序
ordering_genes <- row.names (subset(diff_test_res, qval < 0.01));length(ordering_genes)
## [1] 4617
cds <- setOrderingFilter(cds, ordering_genes)
cds <- reduceDimension(cds, max_components = 2,
method = 'DDRTree')
# 细胞排序
cds <- orderCells(cds)
# 可视化
plot_cell_trajectory(cds, color_by = "Biological_Condition")
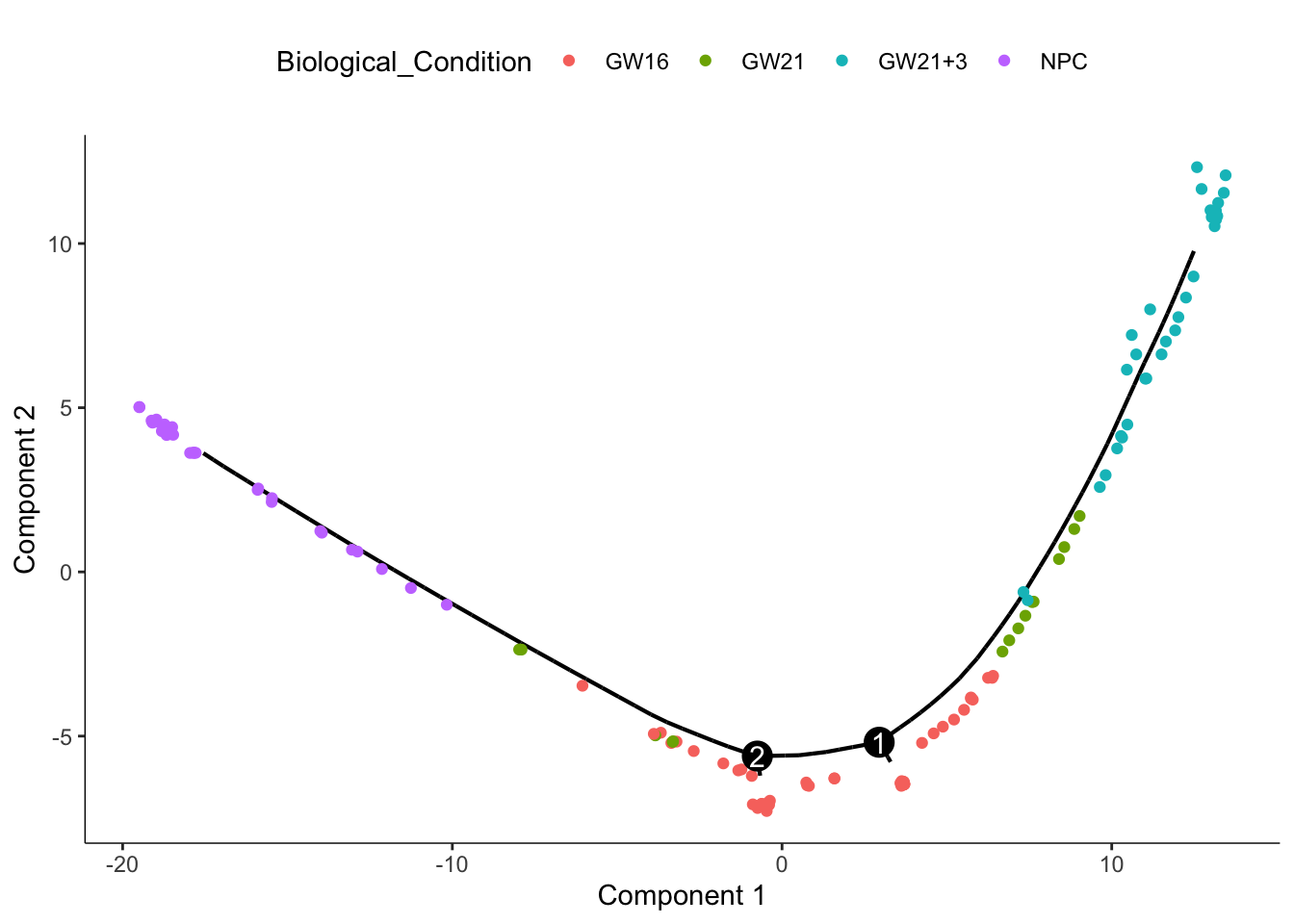
# 展现marker基因
plot_genes_in_pseudotime(cds[cg,],
color_by = "Biological_Condition")


















 1382
1382

 被折叠的 条评论
为什么被折叠?
被折叠的 条评论
为什么被折叠?
 到【灌水乐园】发言
到【灌水乐园】发言











