







1. Monocle3 简介
1.1 基本原理
Monocle3 是一种基于单细胞RNA测序(scRNA-seq)数据的轨迹推断方法,旨在模拟细胞随时间变化的轨迹,推测其潜在的发育或状态转换路径。它通过无监督学习方法,基于细胞的转录组相似性,构建细胞状态的拓扑结构。
1.2 Monocle3 解决的问题
- 解析 细胞状态的连续变化,如细胞分化、激活、耗竭等过程。
- 识别 不同细胞亚群的分化路径,预测其发展方向。
- 通过 轨迹分析 确定关键时间点,以便研究基因动态调控。
1.3 需要的数据
- 单细胞转录组表达矩阵(基因 x 细胞)
- 细胞元数据(细胞类型、分组信息等)
- 基因注释信息(基因名称、ID等)
2. 数据预处理与子集提取
2.1 载入数据与筛选感兴趣的细胞亚群
scRNA1 = readRDS('./tmp/scRNA_harmony_Tcell_annotation.rds')
CD8_tj<-subset(scRNA1, T_cell_annotation %in% c("Cluster_A", "Cluster_B", "Cluster_C"))
- 读取预处理后的单细胞数据。
- 提取感兴趣的细胞亚群进行轨迹分析。
2.2 标准化和降维分析
CD8_tj <- CD8_tj %>%
NormalizeData() %>% # 归一化
FindVariableFeatures() %>% # 筛选高变基因
ScaleData() %>% # 标准化
RunPCA() %>% # PCA降维
RunUMAP(dims = 1:10)
- 进行数据归一化和标准化处理。
- 通过 PCA 降维,提高计算效率。
- 运行 UMAP 进行降维,可视化细胞分布。
3. 构建 Monocle3 CellDataSet
library(monocle3)
mat <- GetAssayData(CD8_tj, slot = "data")
cellInfo <- CD8_tj@meta.data
geneInfo <- data.frame(gene_short_name = rownames(mat), row.names = rownames(mat))
cds <- new_cell_data_set(expression_data = mat,
cell_metadata = cellInfo,
gene_metadata = geneInfo)
- 载入
monocle3包。 - 提取 基因表达矩阵、细胞元数据 和 基因注释信息。
- 创建 Monocle3 轨迹分析所需的
CellDataSet(cds)。
cds <- preprocess_cds(cds, num_dim = 50)
reducedDim(cds, "UMAP") <- Embeddings(CD8_tj, "umap")
- 进行数据预处理,降维并赋予 UMAP 坐标。
4. 轨迹学习与可视化
4.1 轨迹学习
cds = cluster_cells(cds, cluster_method = 'louvain')
cds = learn_graph(cds, use_partition=T, verbose=T, learn_graph_control=list(
minimal_branch_len=10
))
- 进行 细胞聚类(louvain方法)。
- 通过 learn_graph 学习轨迹结构,设定最小分支长度,避免过短分支。
4.2 设定轨迹起始点
start = c("Cluster_A")
closest_vertex = cds@principal_graph_aux[["UMAP"]]$pr_graph_cell_proj_closest_vertex
closest_vertex = as.matrix(closest_vertex[colnames(cds), ])
root_pr_nodes = igraph::V(principal_graph(cds)[["UMAP"]])$name
flag = closest_vertex[as.character(colData(cds)$T_cell_annotation) %in% start,]
flag = as.numeric(names(which.max(table( flag ))))
root_pr_nodes = root_pr_nodes[flag]
cds = order_cells(cds, root_pr_nodes=root_pr_nodes)
- 选择 某个细胞亚群作为轨迹的起点。
- 计算该群体中最近的轨迹节点,重新排序细胞。
4.3 绘制轨迹图
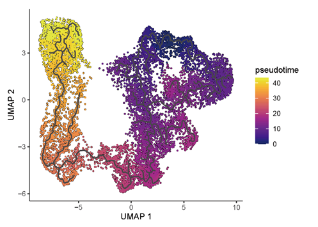
p = plot_cells(cds,
color_cells_by = "pseudotime",
label_cell_groups=F,
label_groups_by_cluster=F,
label_roots=F,
label_leaves=F,
label_branch_points=F,
cell_size=0.5,
group_label_size=3,
rasterize=F)
ggsave(p, filename = './output/Pseudotime/Pseudotime_umap.pdf', width = 5.5, height = 4)
- 以 拟时序(pseudotime) 进行颜色标注。
- 隐藏标签,突出轨迹趋势。
5. 数据导出与保存
5.1 导出拟时序时间信息
time = pseudotime(cds)
time[time==Inf] = 0
time = data.frame(cell=names(time), pseudotime=time)
write.table(time, file="./output/Pseudotime/pseudotime_data.txt", sep="\t", quote=F, col.names=T, row.names=F)
- 计算并导出 拟时序时间数据。
5.2 存储分析结果
saveRDS(cds, file="./tmp/monocle3_result.rds")
saveRDS(CD8_tj, file = "./tmp/seurat_processed_data.rds")
- 保存 Monocle3 轨迹对象 (
cds)。 - 保存 Seurat 处理后的细胞对象 (
CD8_tj)。
总结
本流程基于 Monocle3 构建 细胞轨迹分析,包括:
- 数据预处理(归一化、降维、UMAP)
- 构建 Monocle3 对象(CellDataSet)
- 轨迹学习(cluster_cells, learn_graph)
- 定义轨迹起始点,计算拟时序
- 绘制轨迹图,导出数据
- 结合 RNA velocity 和转录调控分析优化结果


















 3621
3621

 被折叠的 条评论
为什么被折叠?
被折叠的 条评论
为什么被折叠?
 到【灌水乐园】发言
到【灌水乐园】发言











