







 本文介绍了宏基因组测序和宏转录组测序在微生物组学研究中的应用。宏基因组测序能提供微生物群落的物种组成、功能信息,比16S扩增子测序更深入。宏转录组测序则揭示微生物群落的转录活动和调控规律,两者结合可深入理解微生物的功能和响应环境变化的能力。
本文介绍了宏基因组测序和宏转录组测序在微生物组学研究中的应用。宏基因组测序能提供微生物群落的物种组成、功能信息,比16S扩增子测序更深入。宏转录组测序则揭示微生物群落的转录活动和调控规律,两者结合可深入理解微生物的功能和响应环境变化的能力。
原创: 林二狗 宇宙实验媛
宏基因组
宏基因组测序是将环境总DNA提取出来,随机打断成300/500bp的小片段,然后在片段两端加入通用引物进行PCR扩增测序,然后对测序数据进行质控,再将高质量序列拼接,根据数据库参考信息,对基因序列进行预测和功能注释,最终获得重要的宏基因组信息,如序列组成(GC含量、基因组大小等)、物种组成、功能组成和群落特征等。
相比于16S扩增子测序,宏基因组测序能够使物种鉴定深度达到“种”,而前者往往只能达到“属”的级别。另外,基于16S扩增子测序的基因预测结果(预测方法请参见往期内容“微生物组学研究手段概览——扩增子测序”)是依据数据库中的参考序列得到的,宏基因组测序则提供了菌群实际的基因信息。所以,如果不考虑经费的限制,宏基因组测序是能够更准确地研究微生物组及其功能的方法。
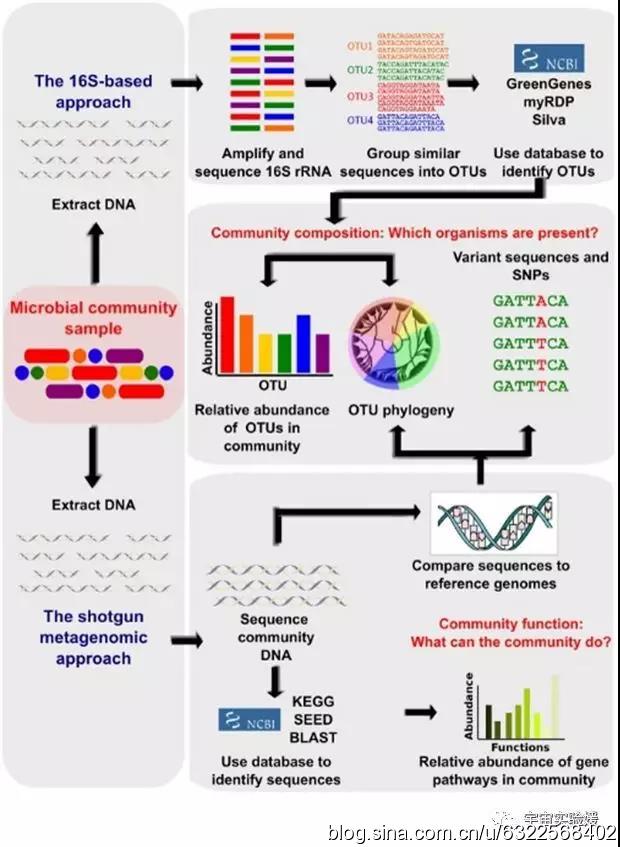
图1. 宏基因组学研究的生物信息工作内容(doi: 10.1371/journal.pcbi.1002808)。
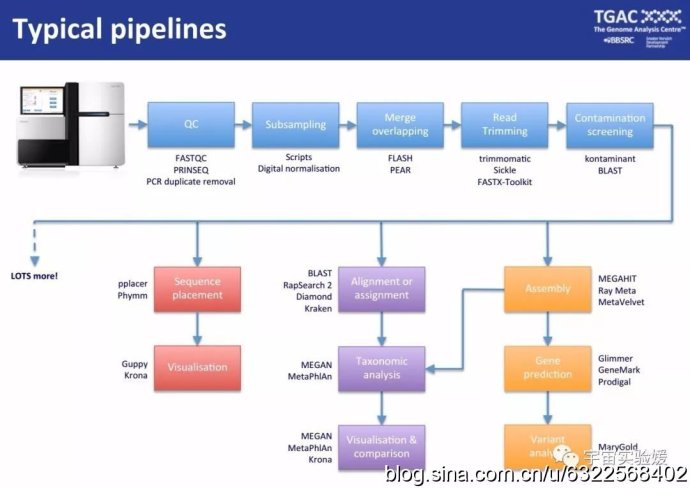
图2. 宏基因组数据处理的基本流程和需用软件(有具体操作需求的同学可以通过下面网址下载英文相关教程https://github.com/TGAC/361Division/tree/master/Metagenomics 2015)
 最低0.47元/天 解锁文章
最低0.47元/天 解锁文章


















 5811
5811

 被折叠的 条评论
为什么被折叠?
被折叠的 条评论
为什么被折叠?
 到【灌水乐园】发言
到【灌水乐园】发言









