







本讲接实战1,本次实战分析主要对 GBM4 样本进行空间区域划分及区域间差异分析
##实战2:空间区域划分及差异分析
##加载R包
library(Seurat)
library(hdf5r)
library(ggplot2)
library(data.table)
library(dplyr)
##载入实战1保存的Seurat对象
load('GBM4.rdata')
#可视化Cluster分布及H&E组织切片
p1<-SpatialPlot(GBM4, label = TRUE, label.size = 5)
p2<-SpatialPlot(GBM4,pt.size.factor = 0.6)+NoLegend()
p1+p2
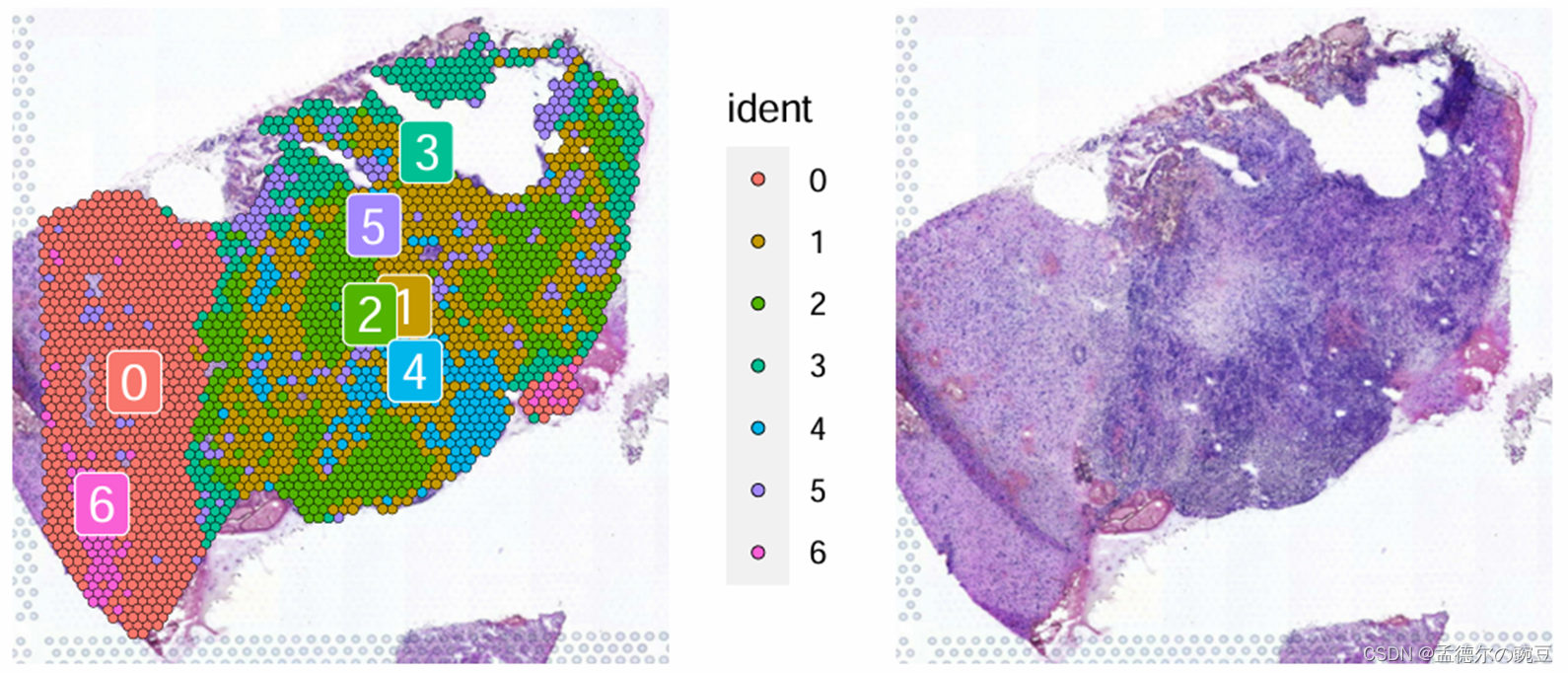
左图:Cluster分布 右图:H&E组织切片
在H&E染色下,癌细胞的细胞核显示出更加浓密和显著的染色,这是由于癌细胞的核质较大、细胞核形态不规则以及核分裂活跃等特点所致。这种深染色现象有助于在组织切片中更容易地识别和定位癌灶。
根据上图(右)H&E 情况,初步将Cluster0 和 6 定义为Normal, 将Cluster3,5定义为Transition, 其他的 Cluster 定义为Tumor。(实际分析中应结合CNV结果,此处仅做示例)
#区域定义
GBM4@meta.data$Region<-NA
GBM4@meta.data$Region[GBM4@meta.data$seurat_clusters %in% c('0','6')] <- "Normal"
GBM4@meta.data$Region[GBM4@meta.data$seurat_clusters %in% c('3','2','5')] <- "Transition"
GBM4@meta.data$Region[GBM4@meta.data$seurat_clusters %in% c('1','4')] <- "Tumor"
#可视化区域划分结果
SpatialPlot(GBM4, label = TRUE, label.size = 5,group.by = 'Region',cols = c('Normal'='#4b5cc4','Transition'='#FE8D3C','Tumor'='#AA0000'))
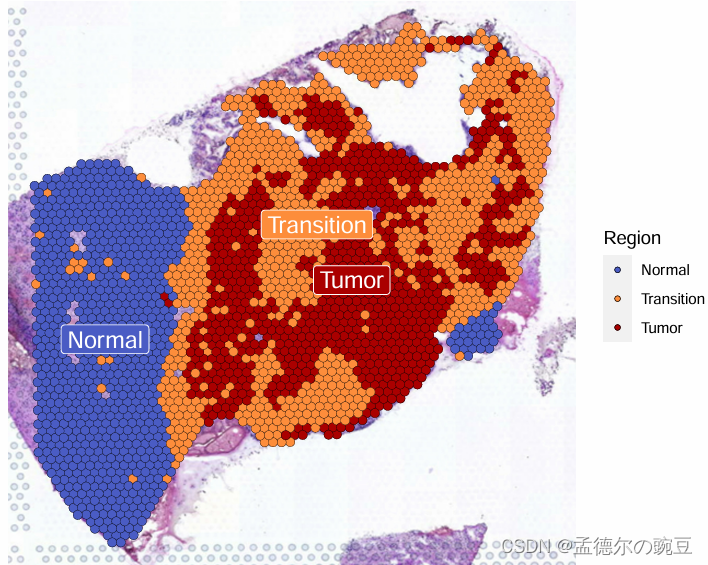
上面根据Cluser及H&E染色特征,大致将切片划分为:Normal、Transition和Tumor区域
接下来就可以对Normal、Transition和Tumor区域进行差异分析了
##区域间差异分析
#切换Idents为上面定义的Region
Idents(GBM4)<-GBM4$Region
#找到各区域的标记物,并只报道阳性位点
markers <- FindAllMarkers(GBM4, only.pos = TRUE, min.pct = 0.25, logfc.threshold = 0.25)
#找到各区域top10基因
top10<-markers %>%
group_by(cluster) %>%
top_n(n = 10, wt = avg_log2FC)
#可视化top10基因热图
DoHeatmap(GBM4, features = top10$gene,group.colors = c('Normal'='#4b5cc4','Transition'='#FE8D3C','Tumor'='#AA0000')) + NoLegend()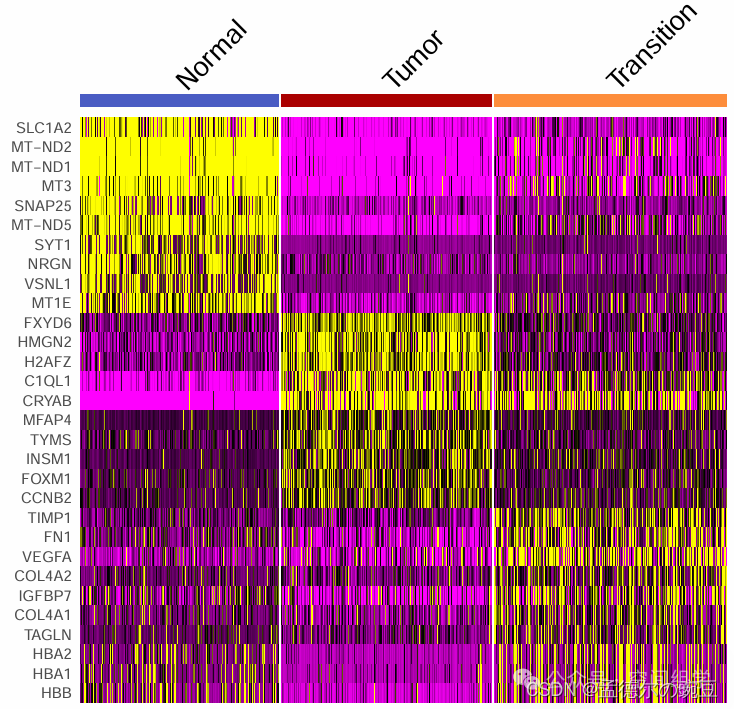
(Normal区域的TOP基因里有几个线粒体,因为未质控,QC是可选项,与单细胞不同)
接下来对Normal、Transition和Tumor区域差异基因进行KEGG富集分析
library(clusterProfiler)
#将基因SYMBOL转换为ENTREZID
gid <- bitr(unique(markers$gene), 'SYMBOL', 'ENTREZID', OrgDb= 'org.Hs.eg.db')
markers <- full_join(markers, gid, by=c('gene' = 'SYMBOL'))
#KEGG通路富集分析
KEGG = compareCluster(ENTREZID ~ cluster, data = markers, fun='enrichKEGG')
#可视化各区域TOP5通路
dotplot(KEGG, label_format=40) + theme(axis.text.x = element_text(angle=45, hjust=1)) + scale_color_gradient(high="#4b5cc4",low="#FE8D3C")
#保存分析结果,以便下期使用
save(GBM4,file= 'GBM4.rdata')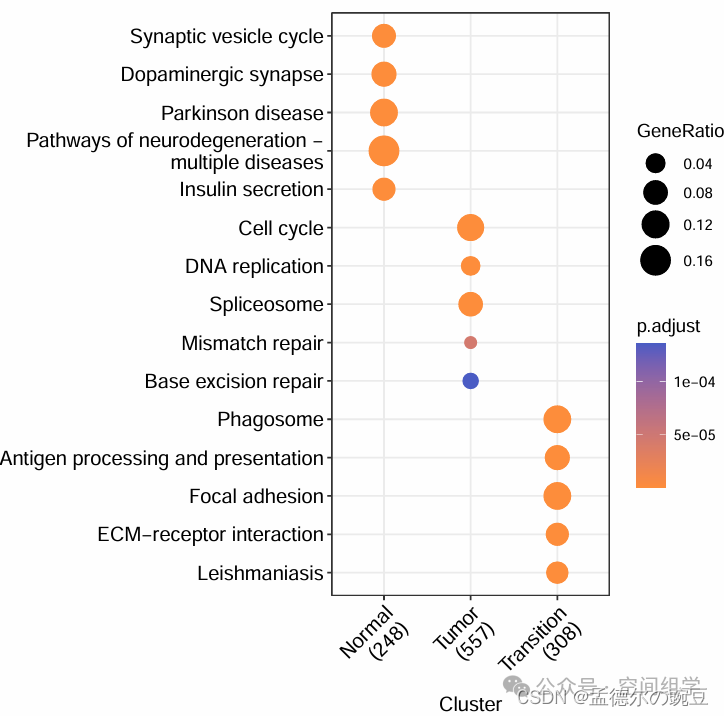
从KEGG富集结果来看,Normal区域、Tumor区域、Transition区域差异显著
Tumor区域的Cell cycle和DNA replication显著富集,肿瘤区域细胞周期和DNA复制的显著富集可能反映了肿瘤细胞在增殖和基因组稳定性方面存在的异常,符合肿瘤的生物学特征;
而Transition区域的Phagosome、Antigen processing and presentation通路富集,表明该区域在机体的免疫防御中起着重要作用;抗原处理和呈递是机体免疫系统中重要的过程,涉及到识别外源抗原或内源抗原,并将它们呈递给免疫系统中的T细胞和B细胞,从而激活免疫反应。
好了,以上就是本次实战2的内容,下期将结合10X单细胞数据对ST空间转录组数据进行反卷积细胞注释!!

















 9万+
9万+

 被折叠的 条评论
为什么被折叠?
被折叠的 条评论
为什么被折叠?
 到【灌水乐园】发言
到【灌水乐园】发言









