







CellChat v2 是一个更新版本,包括:
- 从空间解析的转录组学推断相互作用细胞群之间的空间近端细胞间通讯
- 扩展了数据库 CellChatDB v2,包括 1000 多种蛋白质和非蛋白质相互作用(例如代谢和突触信号转导),并具有丰富的注释。还提供了一个名为的函数,用于轻松更新 CellChatDB
- 新功能可轻松与其他计算工具对接,用于单细胞数据分析和细胞间通信分析
- 交互式 Web 浏览器功能,允许探索空间近端细胞间通信的 CellChat 输出
CellChat V2 空间转录组分析需要四个输入:
-
data.input(基因表达数据 斑点/细胞):基因应与行名和单元格并列 在带有 colnames 的列中。归一化数据(例如,library-size 归一化,然后对数变换,伪计数为 1) 需要作为 CellChat 分析的输入。 -
meta(用户分配的单元格标签和样本 labels):一个数据框(行是带有行名的单元格),包括 的单元格信息,将用于定义单元格组。 -
坐标(空间坐标 斑点/单元格):一个数据框,其中每行都给出空间 每个细胞/点质心的坐标/位置。 -
spatial.factors(空间的空间因素 distance):包含两个距离因子和 的数据框,它依赖于空间 转录组学技术(和特定数据集)。
-
下面开始实战4部分:空间细胞通讯分析
###实战4:空间细胞通讯分析
#安装CellChat v2,从空间转录组学推断相互作用细胞群之间的空间近端细胞间通讯
devtools::install_github("jinworks/CellChat")
##加载R包
library(CellChat)
library(Seurat)
library(tidyverse)
library(patchwork)
#载入实战2保存的 GBM4 空转数据
load('GBM4.rdata')
#可视化Cluster分布
SpatialPlot(GBM4, label = TRUE, label.size = 5)
#CellChat分析中, Cell label不能为‘0’! 因此需手动修改cluster命名
GBM4@meta.data$celltype <- paste("C",GBM4$seurat_clusters,sep = "")
Idents(GBM4) <- "celltype"
SpatialDimPlot(GBM4, label = TRUE, label.size = 5) 
#由于一个spot包含多个细胞,本次使用Cluster进行细胞通讯分析
#获取空转矩阵信息
data.input = Seurat::GetAssayData(GBM4, slot = "data", assay = "SCT")
#获取meta信息
meta = data.frame(labels = Idents(GBM4),
row.names = names(Idents(GBM4)))
unique(meta$labels)
# 获取空间位置信息
spatial.locs = Seurat::GetTissueCoordinates(GBM4, scale = NULL,cols = c("imagerow", "imagecol"))
##scalefactors_json存于GBM4_spaceranger_out/spatial文件夹下
scalefactors = jsonlite::fromJSON(txt = file.path("E:/GSE194329/GBM4_spaceranger_out/spatial", 'scalefactors_json.json'))
spot.size = 65 #10X Visium spot大小为55μm,两个spot之间Gap为10μm
conversion.factor = spot.size/scalefactors$spot_diameter_fullres
spatial.factors = data.frame(ratio = conversion.factor, tol = spot.size/2)
d.spatial <- computeCellDistance(coordinates = spatial.locs, ratio = spatial.factors$ratio, tol = spatial.factors$tol)
#创建CellChat对象
cellchat <- createCellChat(object = data.input,
meta = meta,
group.by = "labels", #定义的名字是labels
datatype = "spatial", #数据类型:空转
coordinates = spatial.locs,
spatial.factors = spatial.factors)
#设置参考数据库
CellChatDB <- CellChatDB.human # use CellChatDB.mouse if running on mouse data
showDatabaseCategory(CellChatDB)
#使用CellChatDB的子集进行细胞间通信分析
CellChatDB.use <- subsetDB(CellChatDB, search = "Secreted Signaling", key = "annotation") #选择Secreted Signaling
cellchat@DB <- CellChatDB.use
##CellChat预处理
cellchat <- subsetData(cellchat) # 即使使用整个数据库,此步骤也是必要的
future::plan("multisession", workers = 4) #多线程
#识别过表达基因
cellchat <- identifyOverExpressedGenes(cellchat)
#识别过表达配体受体对
cellchat <- identifyOverExpressedInteractions(cellchat, variable.both = F)
##细胞间通信网络的推断
cellchat <- computeCommunProb(cellchat, type = "truncatedMean", trim = 0.1,
distance.use = TRUE, interaction.range = 250, scale.distance = 0.01,
contact.dependent = TRUE, contact.range = 100)
参数说明: truncatedMean :用于计算每个细胞组的平均基因表达
distance.use = TRUE:使用空间距离限制作为计算通信概率的约束条件
interaction.range = 250(约4个spot), contact.range = 100(2个spot)
#默认情况下,每个细胞组中用于细胞间通信所需的最小细胞数为10
cellchat <- filterCommunication(cellchat, min.cells = 10)
#在信号通路水平上推断细胞间通讯
cellchat <- computeCommunProbPathway(cellchat)
#计算聚合的 cell-cell 通信网络
cellchat <- aggregateNet(cellchat)
#显示交互次数或总交互次数
groupSize <- as.numeric(table(cellchat@idents))
par(mfrow = c(1,2), xpd=TRUE)
netVisual_circle(cellchat@net$count, vertex.weight = rowSums(cellchat@net$count), weight.scale = T, label.edge= F, title.name = "Number of interactions")
netVisual_circle(cellchat@net$weight, vertex.weight = rowSums(cellchat@net$weight), weight.scale = T, label.edge= F, title.name = "Interaction weights/strength")
#显示celltype间的通讯次数(左)或总通讯强度(右)
p1 <- netVisual_heatmap(cellchat, measure = "count", color.heatmap = "Blues")
p2 <- netVisual_heatmap(cellchat, measure = "weight", color.heatmap = "Blues")
p1 + p2
#展示显著通路结果
cellchat@netP$pathways
#[1] "SPP1" "PTN" "MIF" "MK" "CypA" "IGFBP"
#"ANGPTL"
#[8] "VEGF" "GRN" "PSAP" "SEMA3" "BMP" "FGF"
#"VISFATIN"......
par(mfrow=c(1,1), xpd = TRUE)# xpd = TRUE以显示标题
pathways.show <- c("MIF")
netVisual_aggregate(cellchat, signaling = pathways.show, layout = "circle")
#在空间转录组学上显示信号网络
netVisual_aggregate(cellchat, signaling = pathways.show, layout = "spatial", edge.width.max = 2,vertex.size.max = 1, alpha.image = 0.2, vertex.label.cex = 3.5)
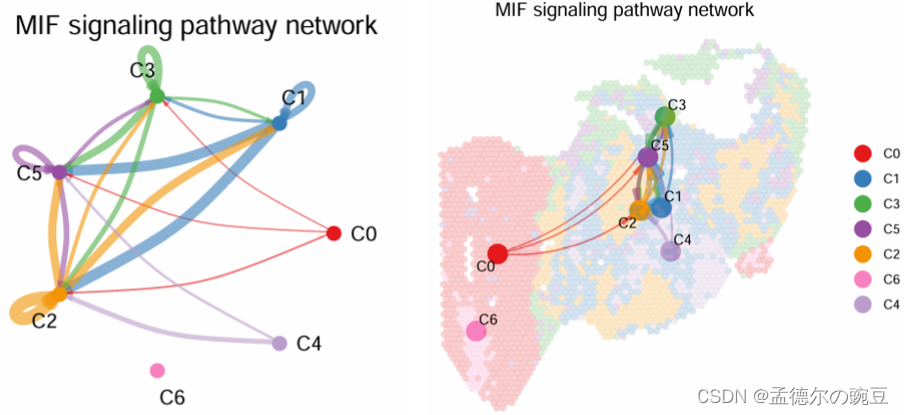
空间水平上显示 MIF 信号网络 (右)
#计算和可视化网络中心性分数:
cellchat <- netAnalysis_computeCentrality(cellchat, slot.name = "netP")#“netP”是指推断的信号通路的细胞间通信网络
par(mfrow=c(1,1))
netAnalysis_signalingRole_network(cellchat, signaling = pathways.show, width = 8, height = 2.5, font.size = 10)

MIF 信号网络中心性分数
#在空间转录组学上显示信号网络时,可以以更大的圆圈表示更大的传入信号
par(mfrow=c(1,1))
netVisual_aggregate(cellchat, signaling = pathways.show, layout = "spatial", edge.width.max = 2, alpha.image = 0.2, vertex.weight = "incoming", vertex.size.max = 4, vertex.label.cex = 3.5)
#取配体-受体对的输入,并以二进制形式显示表达
spatialFeaturePlot(cellchat, pairLR.use = "MDK_SDC1", point.size = 1, do.binary = TRUE, cutoff = 0.05, enriched.only = F, color.heatmap = "Reds", direction = 1)
更大的圆圈表示更大的传入信号(左) ,配体-受体对的空间分布(右)
#可视化组织上的基因表达分布
spatialFeaturePlot(cellchat, features = c("MDK","SDC1"), point.size = 0.8, color.heatmap = "Reds", direction = 1)

#绘制配体受体气泡图,指定受体-配体细胞类型
netVisual_bubble(cellchat, sources.use = c(1),
targets.use = c(3,5), remove.isolate = FALSE)
##保存 CellChat 对象
saveRDS(cellchat,file = 'GBM4-Cellchat.rds')
以上就是本次实战4 CellChat 在空转数据中的应用,更多可视化可自行探索。下期将继续带来一系列空间转录组学分析方法及可视化分享!
完整版代码及更多可视化方式,关注公众号:空间组学 ,回复 cellchat2,自动获取。
















 1400
1400











 被折叠的 条评论
为什么被折叠?
被折叠的 条评论
为什么被折叠?
 到【灌水乐园】发言
到【灌水乐园】发言









