






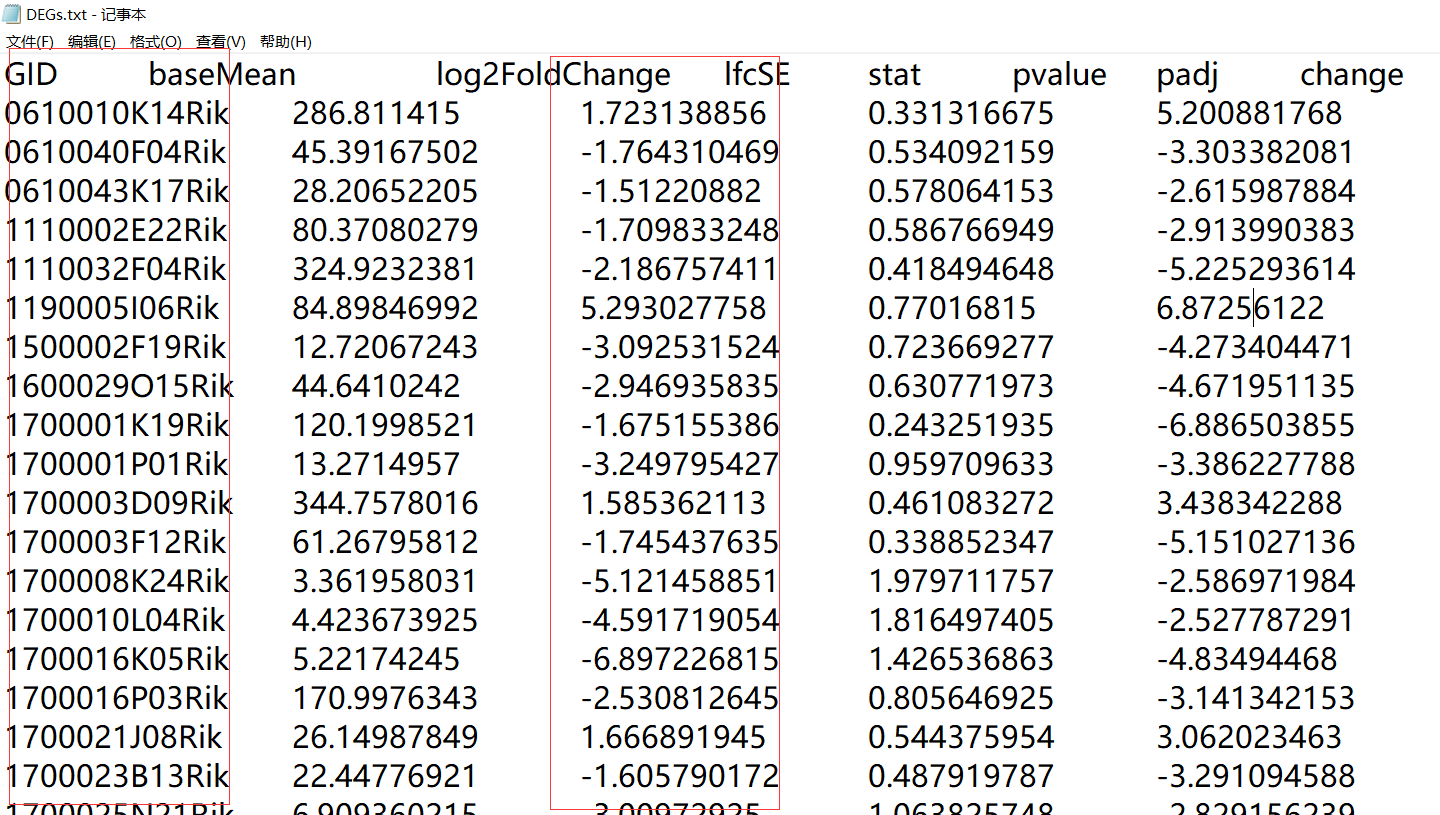
准备好输入文件,输入文件为差异表达分析结果,分析结果主要主要的是gene_ID,和log2foldchange这两列,如图红框框住部分
GSEA输入数据需要将gene根据log2FoldChange值从大到小的顺序以此排列,这时我们在R语言中导入数据的时候处理好就可以了。
#所使用的包
library(org.Mm.eg.db)
library(clusterProfiler)
library(GseaVis)
library(ggplot2)
library(enrichplot)
setwd("D:/data/WYQ/2/New/yongICH_vs_yongshame/GSEA/")
DD<-"DEGs.txt"
DEGs<- read.table(DD, header=TRUE, sep = "\t")
new_DEGs <- DEGs[order(-DEGs$log2FoldChange),]#排序
gene_list <- new_DEGs$log2FoldChange #
names(gene_list) <- new_DEGs$GID#得到一个以geneID为名的log2foldchange列表
3,运行GSEGo程序,这里不将BP CC MF分开
gsego_ALL <- gseGO(
gene_list ,
ont = "ALL",
OrgDb = org.Mm.eg.db,
keyType = "SYMBOL",
exponent = 1,
minGSSize = 10,
maxGSSize = 500,
pvalueCutoff = 0.05,
verbose = FALSE
)
保存结果
ego_results<-as.data.frame(gsego_ALL)
write.csv(ego_results, "GSEA_GO_results.csv")
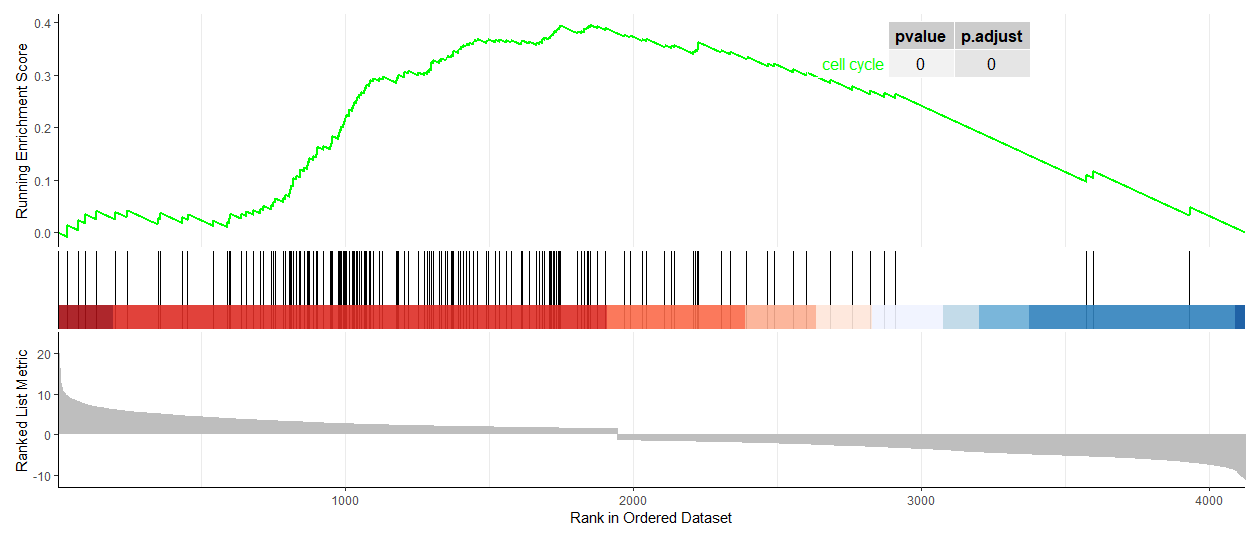
4,可视化其中通路情况,如GO:0000165,GO:0043408这两个通路
paths <- c(
"GO:0000165",)
gseaplot2(gsego_ALL,paths, pvalue_table=TRUE)
5.可视化所有富集结果,并显示上下调通路情况,这里的演示数据只使用了部分数据,所以不全

dotplot(gsego_ALL,showCategory = 40,split=".sign")+facet_wrap(~.sign,scales = "free")















 318
318











 被折叠的 条评论
为什么被折叠?
被折叠的 条评论
为什么被折叠?
 到【灌水乐园】发言
到【灌水乐园】发言









