







两组比较(有重复)
输入数据格式
上游分析进行bismark_methylation_extractor后获得一个包含计数信息的coverage文件,计数文件包含:chr,start,end,methylation%,count methylated,count unmethylated。修改此文件即可。
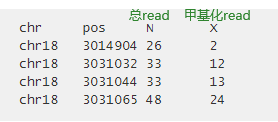
读入输入文件:makeBSsepData
dat.list是一个列表,里面的元素是所有样本的输入文件
BSobj = makeBSsepData(dat.list,
c("C1","C2","N1","N2")) [1:1000,]
call DML:DMLtest + callDML
包括以下步骤:估算所有CpG位点的平均甲基化水平;估算每个CpG位点的离散度;进行Wald-test。
dml = DMLtest(BSobj,group1 = c("C1","C2"),group2 = c("N1","N2"),smoothing = TRUE)
对于WGBS数据,建议进行平滑处理。
使用callDML函数call DML:
dmls = callDML(dml,p.threshold = 0.001,delta = 0.1)
delta指定差异阈值
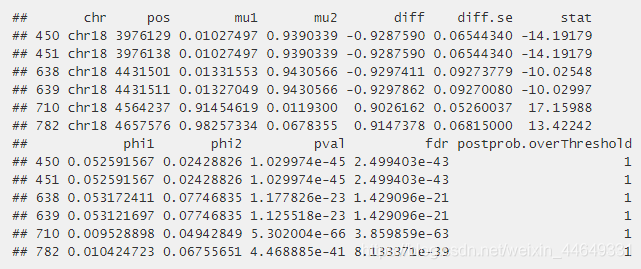
call DMR:callDMR
dmrs = callDMR(dml,p.threshold = 0.05,delta = 0.1)
选择一个合理的阈值来定义DMRs是非常困难的,所以建议尝试不同的阈值。

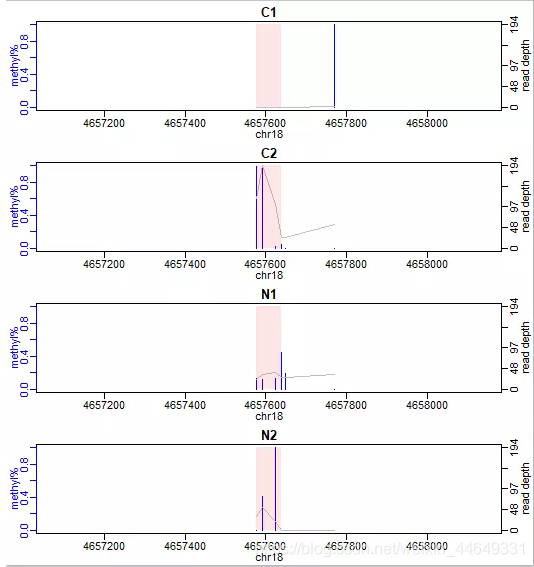
DMR可视化:showOneDMR
showOneDMR(dmrs[1,],BSobj)
第一个甲基化区域
单样本VS单样本
BSobj = makeBSsepData(dat.list,
c("C1","N1")) [1:1000,]
dml = DMLTest(BSobj,group1 = c("C1"),group2 = c("N1"),smoothing = TRUE)
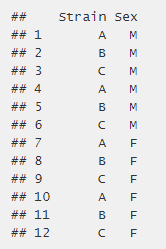
实验设计:两个参数及以上
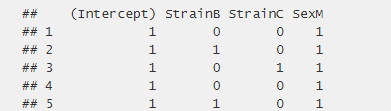
- 假设参数有:菌株种类(A,B,C),性别(M,F)
- 构建design矩阵:
- 建立线性模型:
X = model.matrix(~Strain+ sex,design)
- 使用DMLfit.multiFactor拟合线性模型:
DMLfit = DMLfit.multiFactor(BSobj,design,formula = ~Strain+ sex+Strain:sex)
- 使用DMLtest.multiFactor进行DML test:结果不用再callDML,只需要callDMR。
colnames(DMLfit$X)
dmlTest=DMLtest.multiFactor(DMLfit,coef=2)
coef是指定设计矩阵第n列的参数。这里可以使用term直接指代参数名。














 2288
2288











 被折叠的 条评论
为什么被折叠?
被折叠的 条评论
为什么被折叠?
 到【灌水乐园】发言
到【灌水乐园】发言









