










近十年天然产物药物的生物合成研究进展

摘要
天然产物一直是潜在的先导药物的重要来源,天然产物及其结构类似物在历史上对疾病治疗做出了重大贡献,特别是对癌症和传染病的治疗。在过去两百年的时间里,天然产物的发现和研究经历了巨大的变化,由传统的分离鉴定为主的经典研究方法转为了基因组时代的多学科组合研究。虽然近二十年发现和挖掘了丰富的活性天然产物,但与自然界中巨大的天然产物合成潜力相比仍有不足,庞大的陆地和海洋天然产物资源尚待开发。同时,与传统的化学合成分子相比,天然产物具有丰富的骨架多样性和结构复杂性,在新药发现中展现了巨大的优势。虽然在天然产物的新药创新方面仍面临着种种挑战,但新的分析技术和挖掘策略的出现有望迎来天然产物发现的新阶段。本文总结了近十年(2014年1月—2023年10月)美国食品药品监督管理局批准成药的天然产物及源自天然产物的半合成药物,并对其中纯天然产物来源分子、重要的半合成天然产物前体的生物合成研究进展进行了详细总结。此外还简要总结了一些FDA批准的老药在过去十年中取得的重要生物合成研究进展。期望通过对成药天然产物生物合成途径及机制的深入理解,为更多天然产物创新药物的发现和研究提供借鉴。
长久以来,天然产物一直被认为是治疗性药剂中的重要活性成分。在现代医学出现之前,从植物中提取活性成分并应用于疾病治疗的历史已经长达数千年,比如在我国的典籍《五十二病方》(247种药)、《神农本草经》(365种药)以及《新修本草》(850种药)中,都有大量关于天然产物使用的文献记载[1-2]。经历了漫长的应用历史之后,直到近代,人们开始探究天然药物中真正发挥药效的活性成分,由此逐渐进入了以发现和分离鉴定为主的经典天然产物研究阶段,发现了一系列活性天然产物,如吗啡(morphine)、奎宁(quinine)、麻黄碱(ephedrine)等。进入20世纪之后,活性天然产物的发现突飞猛进,特别是青霉素的发现开启了天然产物抗生素发现的黄金时代。在不到100年的时间里,抗生素彻底改变了现代医学,将人类的平均寿命大大延长[3]。随着天然产物化学技术和生物合成研究的不断进步,以及生物信息学的发展和基因组数据库的丰富,在20世纪末期天然产物挖掘进入了第二个黄金期——基因组时代。一系列新的天然产物发掘策略应运而生,比如激活基因的突变、敲除或过表达,沉默基因的表观遗传操纵、全局调控因子或转录因子过表达、启动子交换以及异源表达策略等[4-5],此外,计算机比对和分析方法的快速发展也大大提高了生物合成基因簇(biosynthetic gene cluster,BGC)的预测准确性,例如基于局部比对算法的搜索工具BLAST(basic local alignment search tool)和隐马尔可夫模型HMM(hidden Markov model)的应用,BGC数据库MIBiG和antiSMASH的建立,以及基于机器学习和深度学习方法的metaBGC和DeepBGC的开发等[6]。人们从基因组数据中预测的生物合成基因簇数量超过100万条,发现了大量具有化学多样性的活性天然产物[7-8]。但是时至今日,对于天然产物的挖掘和利用在天然产物总量中所占的比例仍然非常有限,例如植物中仅有5%~15%进行过系统的天然产物研究,对细菌和真菌而言甚至更低,分别约为1%和5%,此外还有巨大的海洋天然产物资源有待开发[9]。基于天然产物药物发现的内在复杂性,需要高度整合的跨学科方法的应用,而随着现代分子和细胞生物学、分析化学和药理学的进步,具有独特性质的天然产物正在被更多地开发和利用,获得了大量具有临床治疗效果的天然产物分子实体[2,10-12]。
与典型的合成小分子文库相比,天然产物库富含的生物活性化合物覆盖了更广泛的化学空间,它们通常具有更高的分子量,更为丰富的骨架多样性和结构复杂性。此外,天然产物在生物体内的合成通常是用于发挥特定的生物功能,例如参与调节内源性的防御机制以及与其他生物体的生存竞争,这也解释了天然产物与传染病和癌症治疗作用的高度相关性[13-14]。但是,天然产物对药物研发的贡献也面临挑战,例如新颖活性分子的有效挖掘、避免已知化合物的重新发现、具有生物活性的微量天然产物的分离和表征、用于探索构效关系的天然产物类似物的生成等等,此外,复杂天然药物在工业生产方面也存在诸多瓶颈,大多数药用天然产物在自然界中的含量低,化合全合成难度极大,生产成本的增加严重限制了天然产物药物的临床应用,这也使制药工业从20世纪90年代起减少了对天然产物的研发动力[15]。近年来,随着科学技术的不断发展,包括分析工具的改进、基因组挖掘和工程策略的持续升级、微生物培养的进步,正在逐步解决这些困难并开辟新的机会,实现了越来越多的复杂天然产物的工业全合成,如艾拉环素、奥利司他等[16-18]。因此,对天然产物作为先导药物的兴趣正在恢复,特别是在耐药菌抗生素的发展方面[13,19-22]。
本文系统总结了近十年来由美国食品药品监督管理局(Food and Drug Administration,FDA)批准的天然产物药物及天然产物半合成药物,着重介绍了完全来源于天然产物的药物分子和重要的半合成药物前体的生物合成研究进展,涵盖了类固醇、聚酮及脂肪酸类、非核糖体肽类、生物碱类及萜类等多个不同类型的天然产物,此外也简要汇总了一些更早被FDA批准的药物在近十年取得的重大生物合成进展。在这些天然产物药物或其类似物的生物合成研究中取得的最新进展,为药物分子的进一步优化和创新提供了宝贵借鉴,同时对于未来新药的发现也具有指导意义。
1 近十年批准的天然产物药物及天然产物半合成药物简介
在过往FDA批准的新药中,以天然产物或天然产物半合成衍生物为新分子实体的药物是其中的重要组成部分。本文根据FDA数据库(https://www.accessdata.fda.gov/scripts/cder/daf/index.cfm)检索整理了近十年(2014年1月到2023年10月)获得批准的天然产物药物及天然产物半合成药物,并对这些药物的生物合成进展进行了归纳总结(表1和图1)。
表1 FDA批准的天然产物药物和半合成药物总结
Table 1 Summary of FDA-approved natural product drugs and semisynthetic drugs
| 批准药物 | 药物前体 | 来源 | 主要功效 | 结构类型 | 批准时间 | 商品名 | 参考文献 |
|---|---|---|---|---|---|---|---|
| Natural product drugs: | |||||||
| 牛磺酸二醇(taurursodiol)* | — | Ursusthibetanus | antiapoptotic | steroid | 2022 | Relyvrio | [23] |
| tapinarof* | — | Photorhabdusluminescens | AhR agonist | polyketide | 2022 | Vtama | [24] |
| 雌四醇(estetrol)* | — | humans | hormone regulation | steroid | 2021 | Nextstellis | [25] |
| 大麻二酚(cannabidiol)* | — | Cannabissativa L. | analgesic, anticonvulsant | polyketide | 2018 | Epidiolex | [26] |
| 血管紧张素(angiotensin)Ⅱ* | — | humans | blood pressure regulation | peptide | 2017 | Giapreza | [27] |
| 普拉睾酮(prasterone)* | — | humans | hormone regulation | steroid | 2016 | Intrarosa | [28] |
| 曲贝替定(trabectedin)* | — | CandidatusEndoecteinascidiafrumentensis | antitumor | non-ribosomal peptide | 2015 | Yondelis | [29] |
| 胆酸(cholic acid)* | — | animals | facilitating fat absorption | steroid | 2015 | Cholbam | [30] |
| 去氧胆酸 (deoxycholic acid)* | — | animals | cytolytic agent | steroid | 2015 | Kybella | [30] |
| Semisynthetic drugs: | |||||||
| 瑞扎芬净(rezafungin)* | echinocandin | Aspergillusdelacroxii | antifungal | non-ribosomal peptide | 2023 | Rezzayo | [31] |
| 艾瑞芬净(ibrexafungerp) | enfumafungin | Hormonemacarpetanum | antifungal | terpene | 2021 | Brexafemme | [32] |
| 伏环孢素(voclosporin) | cyclosporine | Beauverianivea | calcineurin inhibitor | non-ribosomal peptide | 2021 | Lupkynis | [33] |
| 克拉司酮(clascoterone) | progesterone | humans | topical androgen antagonist | steroid | 2020 | Winlevi | [34] |
| 青蒿琥酯(artesunate) | artemisinin | Artemisiaannua | antimalarial | terpene | 2020 | Artesunate | [35] |
| 乳糖醇(lactitol) | lactose | animals | osmotic laxative | sugar alcohol | 2020 | Pizensy | [36] |
| 来法莫林(lefamulin)* | pleuromutilin | Clitopiluspasseckerianus | antibacterial | terpene | 2019 | Xenleta | [37] |
| 布瑞诺龙(brexanolone) | pregnanolone | humans | antidepressant | steroid | 2019 | Zulresso | [38] |
| 利福霉素 (rifamycin)SV | rifamycin B | Amycolatopsisrifamycinica | antibacterial | polyketide | 2018 | Aemcolo | [39] |
| 奥玛环素(omadacycline) | tetracyclines | Streptomyces | antibacterial | polyketide | 2018 | Nuzyra | [40] |
| 普拉佐米星(plazomicin) | sisomicin | Micromonosporainositola | antibacterial | aminoglycoside | 2018 | Zemdri | [41] |
| 莫昔克丁(Moxidectin) | avermectin | Streptomycesavermitilis | antiparasitic | polyketide | 2018 | Moxidectin | [42] |
| 艾拉环素(eravacycline) | tetracyclines | Streptomyces | antibacterial | polyketide | 2018 | Xerava | [43] |
| 萨瑞环素(sarecycline) | tetracyclines | Streptomyces | antibacterial | polyketide | 2018 | Seysara | [44] |
| 米哚妥林(midostaurin) | staurosporine | Streptomycesstaurosporeus | antineoplastic | alkaloid | 2017 | Rydapt | [45] |
| 奥贝胆酸 (obeticholic acid) | cholic acid | animals | FXR agonist | steroid | 2016 | Ocaliva | [46] |
| 尿苷三乙酸酯 (uridine triacetate) | uridine | animals | antidote | nucleoside | 2015 | Xuriden | [47] |
| 头孢洛扎(ceftolozane) | cephalosporin | Acremonium | antibacterial | beta-lactam | 2014 | Zerbaxa | [48] |
| 奥利万星(oritavancin) | chloroeremomycin | Amycolatopsisorientalis | antibacterial | glycopeptide | 2014 | Orbactiv | [49] |
| Earlier FDA-approved drugs: | |||||||
| 奥利司他(orlistat)* | lipstatin | Streptomycestoxytricini | lipase inhibitor | fatty acid | 1999 | Xenical | [50] |
| 依托泊苷(etoposide)* | podophyllotoxin | Podophyllumhexandrum | antitumor | lignan | 1983 | Vepesid | [51] |
| 长春花碱(vinblastine)* | — | Catharanthusroseus | antitumor | alkaloid | 1965 | Velban | [52] |
| 长春新碱(vincristine)* | — | Catharanthusroseus | antitumor | alkaloid | 1963 | Oncovin | [52] |
| 秋水仙碱(colchicine)* | — | Gloriosasuperba | anti-inflammatory | alkaloid | 1961 | Colbenemid | [53] |
| 吗啡(morphine)* | — | Papaversomniferum | analgesic | alkaloid | 1941 | Morphine sulfate | [54] |
| 可待因(codeine)* | — | Papaversomniferum | analgesic | alkaloid | 1950 | Codeine sulfate | [54] |
| 东莨菪碱(scopolamine)* | — | Solanaceae | anticholinergic | alkaloid | 1979 | Transderm Scop | [55] |
| 维生素(vitamin)B12* | — | Archaea, bacteria | supplement | alkaloid | 1942 | Cyanocobalamin | [56] |
| 紫杉醇(paclitaxel)* | — | Taxusbrevifolia | anticancer | terpene | 1992 | Taxol | [57] |
注:包括近十年批准的新药以及近十年取得重大生物合成研究进展的老药;* 为本文总结了生物合成研究进展的药物。
图1
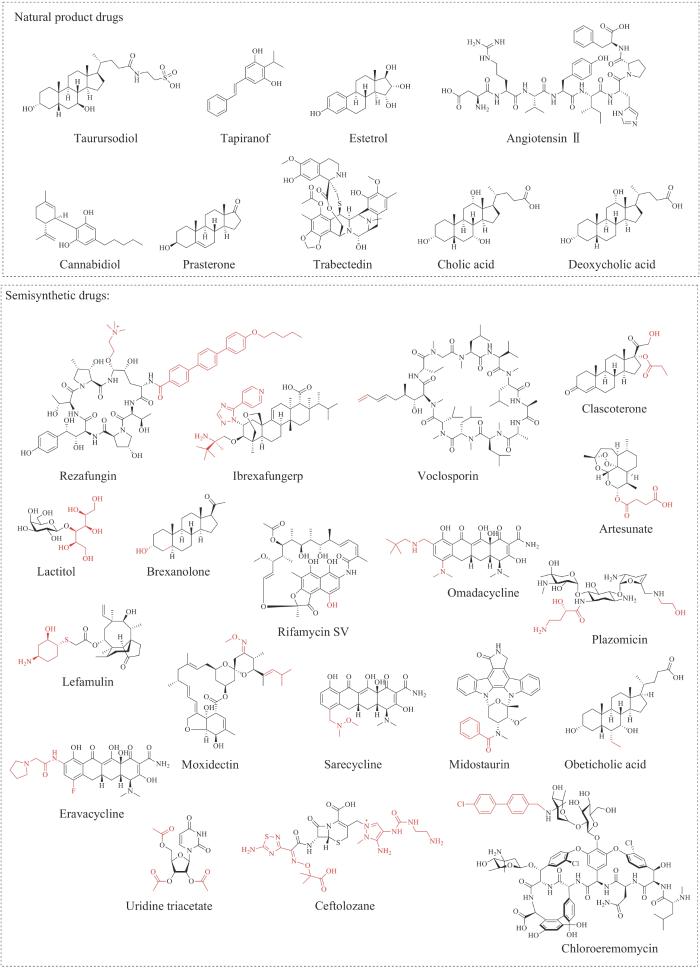
图1 近十年FDA批准的天然产物药物及半合成药物的化学结构
来源于天然产物及天然产物半合成衍生物的新分子实体数量有28例,占FDA批准新药总量(331例)的8.5%,其中,结构完全未改变的天然产物新分子实体有9例,主要来源于人体及动物的类固醇和肽类天然产物,包括改善胆汁酸合成障碍的药物胆酸(cholic acid)、破坏脂肪细胞的药物去氧胆酸(deoxycholic acid)、治疗肌萎缩性侧索硬化症的药物牛磺酸二醇(taurursodiol)、治疗更年期症状的药物普拉睾酮(prasterone)、避孕药物雌四醇(estetrol)和调节血压的药物血管紧张素Ⅱ(angiotensinⅡ),另一部分来源于植物和微生物次级代谢相关的聚酮类和非核糖体肽类天然产物,包括治疗银屑病的药物tapinarof、治疗癫痫的药物大麻二酚(cannabidiol)和抗肿瘤药物曲贝替定(trabectedin)。由天然产物来源的前体分子半合成创制的新分子实体有19例,包括非核糖体肽类,如抗真菌药物瑞扎芬净(rezafungin);萜类,如抗革兰氏细菌药物来法莫林(lefamulin);聚酮类,如抗寄生虫药物莫昔克丁(moxidectin)等。总体而言,FDA平均每年批准的天然产物药物和天然产物半合成药物数量较为稳定,占药物总量的比例也相对一致。
此外,FDA批准的老药里也有一部分药物在最近十年的生物合成研究方面取得了重大进展,不仅在生物合成途径解析方面逐渐完善,包括减肥药物奥利司他(orlistat),抗癌药物依托泊苷(etoposide)、长春花碱(vinblastine)、长春新碱(vincristine),痛风治疗药物秋水仙碱(colchicine),镇痛药物吗啡和可待因(codeine)等,也在工程化微生物细胞中的生物全合成方面取得了重要进展,如抗胆碱药物东莨菪碱(scopolamine)、抗癌药物紫杉醇(paclitaxel)、长春花碱以及维生素B12等(图2)。下文以药物结构类型分类,介绍了上述新、老两类天然产物药物的生物合成研究进展,尤其对结构复杂的植物和微生物来源的天然产物,在生物合成途径及关键酶催化机制上进行了详细的总结。
图2
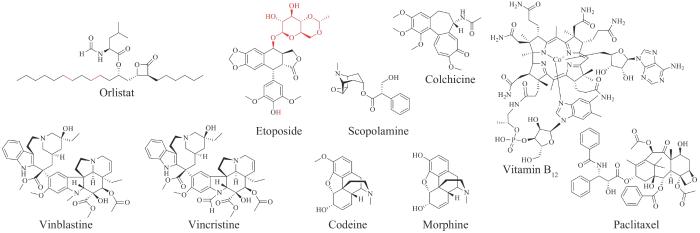
图2 近十年取得重要生物合成进展的FDA批准的老药的化学结构
2 类固醇天然产物
2.1 胆酸和去氧胆酸
动物胆汁在过往的数千年中一直被作为具有临床应用价值的药物,其中主要的天然产物和活性成分为胆汁酸。胆汁酸能够促进脂质(包括脂溶性维生素)的吸收,起到乳化剂的作用,在肠道代谢、细胞信号传导和维持微生物群系组成上具有重要作用[58]。胆汁酸在体内的稳态平衡与人体的健康或疾病息息相关,例如去氧胆酸(deoxycholic acid,DCA)在肠内水平的非正常性升高可诱发肠道紊乱及相关疾病。2015年FDA同时首次批准胆酸(cholic acid,CA)和去氧胆酸分别作为主要活性成分的药物。以胆酸为主要成分的处方药物Cholbam用于治疗胆汁酸合成障碍,以及用于辅助治疗肝脏功能紊乱的过氧化物酶体疾病,如齐薇格(Zellweger)综合征。以去氧胆酸为主要成分的处方药物Kybella用于改善成人下巴以下中度至重度脂肪(也称为双下巴)的外观和轮廓,是目前唯一获得FDA批准的可以破坏脂肪细胞,逐渐改善颏下脂肪外观的可注射药物。
胆汁酸类代谢产物是由24个碳原子组成的两亲性分子,主要包括疏水性的类固醇母核结构,包括三个六元环和一个五元环,附着在其上的亲水性羟基官能团以及柔性的酸性脂肪侧链(图3)[30]。胆汁酸可以分为两大类,以胆固醇为原料直接在肝细胞中合成的胆汁酸称为初级胆汁酸,主要包括胆酸、鹅去氧胆酸(chenodeoxycholic acid,CDCA)以及其他一些在C24位结合牛磺酸(taurine)或甘氨酸的衍生物。初级胆汁酸在肠道中被大量肠内微生物代谢修饰(如7α-脱羟基反应、CA或CDCA的去结合反应)之后产生的胆汁酸称为次级胆汁酸,如DCA、石胆酸(lithocholic acid,LCA)等,次级胆汁酸是具有更广泛活性和功能的代谢产物。按照胆汁酸的结构特点可以将其分为游离型胆汁酸和结合型胆汁酸,传统认为结合型胆汁酸中氨基酸的结合主要发生在肝细胞中,由胆酸辅酶A:氨基酸N-酰基转移酶(bile acid-CoA: amino acid N-acyltransferase,BAT)负责酰基偶联,所产生的结合型胆汁酸分泌到肠道中进行代谢吸收[59]。
图3
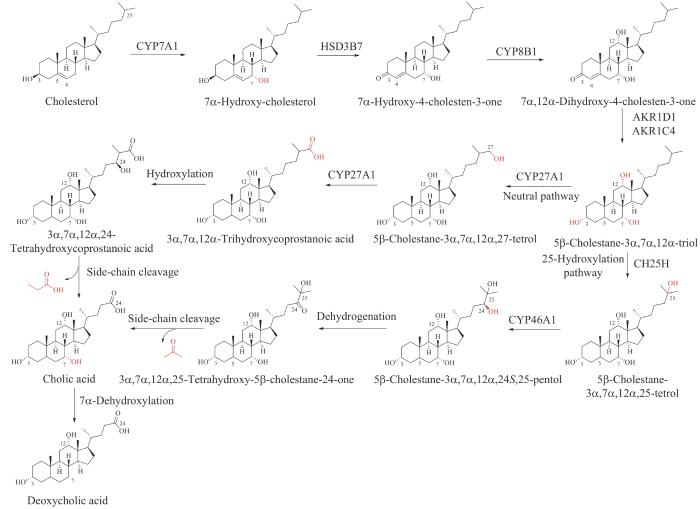
图3 胆酸和去氧胆酸的生物合成
CYP7A1—胆固醇7α-羟化酶;HSD3B7—3β-羟基-Δ5-C27-类固醇氧化还原酶;CYP8B1—12α-羟化酶;AKR1D1—Δ5-3-氧代类固醇5β-还原酶;AKR1C4—3α-羟基类固醇脱氢酶;CYP27A1—甾醇27-羟化酶;CH25H—甾醇25-羟化酶;CYP46A1—24β-羟化酶
胆汁酸类代谢产物的生物合成通常发生在内质网、线粒体、细胞质和过氧化物酶体中,其生物合成路径涉及4条不同的途径,包括中性途径(或经典途径)、酸性途径(或替代途径)、Yamasaki途径以及25-羟化途径,其中中性途径占胆汁酸类代谢产物合成总量的90%,4条途径所包含的反应类型包括类固醇母核的修饰(如羟基化、C5-C6双键的还原、C3位羟基差向异构化)、侧链的脱除以及与甘氨酸和牛磺酸的结合,通常类固醇母核的修饰发生在侧链脱除之前[30]。中性途径和酸性途径发生在肝脏细胞中。首先,胆固醇7α-羟化酶催化胆固醇的C7位发生羟基化产生7α-羟基胆固醇,该步骤为胆汁酸合成的限速步骤,然后7α-羟基胆固醇经一系列的环修饰步骤得到5β-胆甾烷-3α,7α,12α-三醇(5β-cholestane-3α,7α,12α- triol),其侧链可被线粒体甾醇27-羟化酶修饰(即中性途径)或微粒体甾醇25-羟化酶修饰(即25-羟化途径)发生羟基化反应。在中性途径中,5β-胆甾烷-3α,7α,12α,27-四醇(5β-cholestane-3α,7α,12α,27-tetrol)通过相同的线粒体27-羟化酶或胞质醇醛脱氢酶氧化为3α,7α,12α-三羟基前列腺酸(3α,7α,12α-trihydroxycoprostanoic acid),最后通过裂解末端三个碳的丙酸分子后转化为胆酸。在25-羟化途径中,5β-胆甾烷-3α,7α,12α,25-四醇的C24位点被微粒体酶进一步羟基化,然后胞质部分裂解5β-胆甾烷-3α,7α,12α,24S,25-五醇(5β- cholestane-3α,7α,12α,24S,25-pentol)释放末端丙酮分子产生胆酸[30,60]。最后,胆酸发生C7位脱羟基化生成去氧胆酸(图3)[61-62]。
2.2 牛磺酸二醇
牛磺酸二醇(taurursodiol,TURSO,又名ursodoxicoltaurine或牛磺熊去氧胆酸,tauroursod- eoxycholic acid,TUDCA)是FDA在2022年9月批准的药物Relyvrio中的活性成分,该药用于治疗肌萎缩性侧索硬化症(也称为渐冻症)。TUDCA是熊去氧胆酸(ursodeoxycholic acid)与牛磺酸的结合物(图4),是一种高度亲水的胆酸,能够减少小肠对胆固醇的吸收,从而降低人体膳食中胆固醇的摄入量和体内胆固醇的含量。同时,TUDCA也具有抗细胞凋亡和内质网应激反应抑制作用,在一些国家用于治疗胆结石,具有较好的疗效和安全性[23,63-65]。
图4

图4 TUDCA在体内及体外的生物合成途径
TUDCA在人体内的含量较低,但在亚洲黑熊(Ursusthibetanus)的体内含量较高,这是目前唯一已知的可大量产生TUDCA的哺乳动物。牛磺鹅去氧胆酸(taurochenodeoxycholic acid,TCDCA)是TUDCA的合成前体,二者构成一对同分异构体。在生物体内,TUDCA是由TCDCA经肠道微生物中的五个催化酶作用而产生。首先,TCDCA由胆盐水解酶(bile salt hydrolase,BSH)催化去结合牛磺酸产生CDCA,然后由短链脱氢酶/还原酶超家族的羟基类固醇脱氢酶(hydroxysteroid dehydrogenase,HSDH)7α-HSDH和7β-HSDH经两步反应产生差向异构产物UDCA,最后经胆酸CoA连接酶(bile acid CoA ligase,BAL)和BAT催化重新结合牛磺酸得到TUDCA[66-69]。而在生物体外,TUDCA可由TCDCA经两步酶催化反应直接合成。首先,TCDCA在辅因子NAD(P)+参与下经7α-HSDH氧化为牛磺酸-7-酮基鹅去氧胆酸(taurine-7-ketolithocholic acid,T7-KLCA),然后在辅因子NAD(P)H参与下经7β-HSDH催化还原生成TUDCA(图4)[70-71]。因此,参与TUDCA生物合成的7α-HSDH和7β-HSDH是其中关键的两个酶。目前,7α-HSDH的编码基因可在多种微生物中克隆得到,如真杆菌(Eubacterium sp.)、大肠杆菌(Escherichiacoli)、梭状芽孢杆菌(Clostridiumsordellii,C.absonum)以及脆弱拟杆菌(Bacteroidesfragilis),7β-HSDH的编码基因也可在梭状芽孢杆菌(C.absonum)、产气柯林斯菌(Collinsellaaerofaciens)以及瘤胃球菌(Ruminococcusgnavus,R.torques)中克隆得到[71]。
2017年王伯初课题组和谭君课题组[71]合作进一步拓展了针对新的TUDCA生物合成关键酶HSDH的挖掘。研究人员通过宏基因组分析策略,在亚洲黑熊的肠道微生物中鉴定得到5个新的7α-HSDH基因和1个新的7β-HSDH基因,并通过体外酶活分析表征了其催化功能,其中4个酶表现出了很好的催化效率。研究数据也表明,黑熊肠道菌群在TUDCA的生物转化中存在合作。该研究为TUDCA的生物合成研究以及胆酸化合物在C7位的差向异构化反应提供了更多具有应用前景的候选催化酶。近些年,TUDCA的生物合成已在异源宿主大肠杆菌及酿酒酵母(Saccharomycescerevisiae)中实现,此外利用7α-HSDH和7β- HSDH双酶固定化耦合系统也实现了TUDCA的体外酶法高效制备[70,72-73]。
值得一提的是,对TUDCA在体内的生物前体UDCA目前已经进行了大量的生物合成和生物转化研究,生物合成UDCA的酶催化或全细胞合成的底物通常为廉价易得的CDCA或者LCA(图5)[74]。许建和课题组[75-77]通过对限制产率的关键酶7β-HSDH的定向进化以及7α-HSDH和7β-HSDH的双酶固定串联方法,实现了从CDCA到UDCA的大规模生物转化。随后该研究团队通过进一步对7β-HSDH的理性设计,改变了7β-HSDH的辅因子偏好性,由严格依赖NADPH转变为可以高效利用更为廉价的NADH。2021年Bornscheuer课题组[78]通过对链霉菌来源的细胞色素P450单加氧酶CYP107D1(OleP)的工程化改造,改变了OleP对LCA的立体和区域选择性,将原本催化LCA产生鼠脱氧胆酸(murideoxycholic acid,MDCA)的6β-羟基化反应变为产生UDCA的7β-羟基化反应。可见通过更多简洁高效的UDCA的生物转化反应的建立,有助于将来应用于TUDCA的体外生物合成研究。
图5
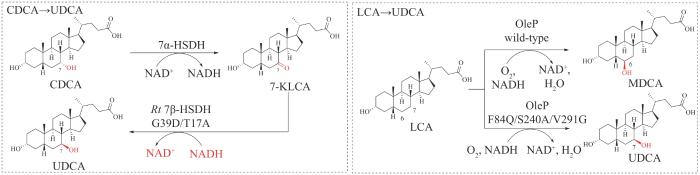
图5 从CDCA和LCA分别合成UDCA的体外生物转化
2.3 普拉睾酮和雌四醇
天然或合成的类固醇激素具有广泛的药物用途,包括作为激素避孕药物的使用以及用于更年期症状的治疗。2016年FDA批准了以普拉睾酮(prasterone)为主要活性成分的药物Intrarosa,用于治疗绝经后妇女的中度至重度外阴和阴道萎缩症状。2021年FDA批准了含有屈螺酮(drospirenone)和雌四醇(estetrol,E4)的组合口服避孕药Nextstellis,是雌四醇作为新的分子实体首次成药,也是世界上第一个获得批准的雌四醇产品,它的安全性和有效性超过了97%的含有合成雌激素乙炔雌二醇(ethinylestradiol,EE2)的口服避孕药,减少了环境中有害的干扰内分泌的化学物质积累[79]。
普拉睾酮也称为脱氢表雄酮(dehydroepia- ndrosterone,DHEA),是一种由肾上腺皮质产生的C19类固醇,在睾丸和卵巢中也有少量产生。DHEA的合成起源于胆固醇,大多数DHEA在分泌前发生硫酸酯化形成DHEA硫酸盐(DHEA sulfate,DHEAS)的形式[28,80-81]。雌四醇发现于20世纪60年代中期,是已知的人体4种天然类固醇雌激素之一[其他3种为雌素酮(estrone,E1)、雌二醇(estradiol,E2)、雌三醇(estriol,E3)]。雌四醇仅在人类妊娠期间由胎儿肝脏合成,并通过胎盘进入母体循环[82]。目前认为雌四醇的合成存在两条途径,第一条途径是在胎盘中形成的雌二醇进入胎儿隔室,首先被迅速硫酸酯化,然后雌二醇硫酸盐在胎儿肝脏中发生15α-/16α-羟基化,从而形成雌四醇硫酸盐,雌四醇硫酸盐水解得到雌四醇(图6,Pathway 1)。同时,也存在另一条重要途径,由胎儿肝脏中形成的15α,16α-二羟基- DHEAS在胎盘中通过水解、脱氢、异构化以及芳构化反应转化为雌四醇,最终通过两条不同的生物合成路径实现雌四醇在人体内的生物合成(图6,Pathway 2)[25,83]。
图6
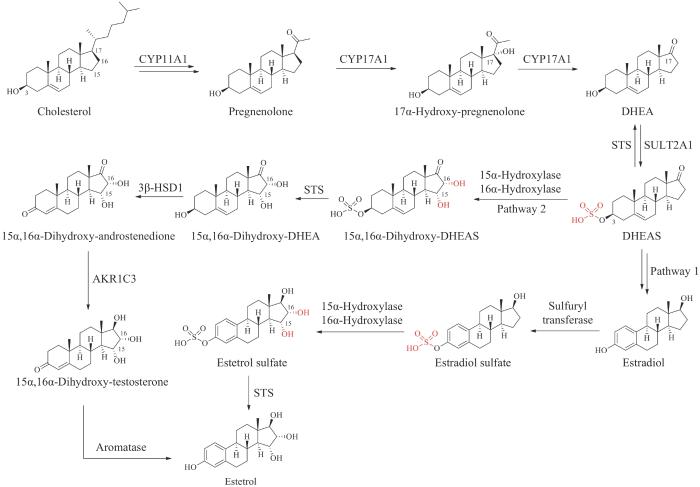
图6 普拉睾酮和雌四醇在人体内的生物合成路径CYP11A1—细胞色素P450胆固醇侧链裂解酶;CYP17A1—17α-羟化酶/17,20-裂解酶;SULT2A1—磺基转移酶;STS—类固醇硫酸酯酶;AKR1C3—醛酮还原酶
3 聚酮及脂肪酸类天然产物
3.1 tapinarof
tapinarof也称为本维莫德(benvitimod),是2022年FDA批准的新药Vtama的主要活性成分,它是一种新型的小分子芳香烃受体激动剂,主要用于成人斑块型银屑病的局部治疗以及特应性皮炎。Vtama是同类产品中第一个也是唯一一个获得FDA批准的不含类固醇的外用药物[84-86]。
tapiranof首次发现是在与异小杆属(Heterorhabditis)线虫共生的革兰氏阴性菌发光杆菌(Photorhabdusluminescens)的代谢物中。1959年研究人员发现含有大量tapiranof的异小杆属线虫在死亡之后不会迅速腐烂,提示其具有潜在的抗菌活性。同时,tapiranof也是一种毒力因子,它能抑制昆虫先天免疫反应的主要组成成分酚氧化酶的活性[87-89]。tapiranof属于二苯乙烯类天然产物,该类化合物广泛分布在植物中,其生物合成途径起源于植物中的Ⅲ型聚酮合酶二苯乙烯合酶(stilbene synthase,STS)催化肉桂酰辅酶A(cinnamoyl-CoA)或者香豆酰辅酶A(coumaroyl-CoA)与3分子丙二酰辅酶A(malonyl-CoA)延伸单元的缩合(图7),然而tapiranof是在非植物范围内首次发现的由革兰氏阴性菌产生的二苯乙烯类天然产物,其C2位连有一个异丙基官能团(图7)。
图7
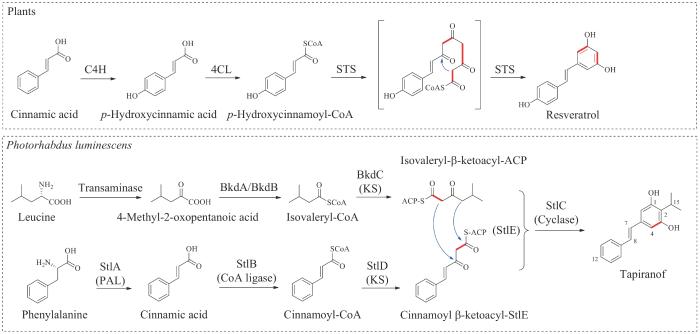
图7 二苯乙烯类天然产物在植物和发光杆菌中的生物合成途径
C4H—肉桂酸-4-羟化酶;4CL—4-香豆酰-CoA连接酶;STS—二苯乙烯合酶;KS—酮基合酶;PAL—苯丙氨酸解氨酶
2008年Bode课题组[24]探究了细菌来源的tapiranof(或异丙基二苯乙烯,isopropylstilbene)特殊的生物合成途径。与植物中的生物合成途径类似的是,tapiranof的合成前体同样起源于肉桂酸(cinnamic acid)。通过同位素喂养及体内基因敲除验证,研究人员在发光杆菌的基因组中鉴定了与tapiranof生物合成相关的独立基因或者操纵子,提出了tapiranof在细菌中可能的生物合成途径(图7)。首先,苯丙氨酸经苯丙氨酸解氨酶和CoA连接酶的连续催化生成肉桂酰辅酶A,1分子的丙二酰辅酶A通过酰基转移酶上载到酰基载体蛋白(acyl carrier protein, ACP)StlE上,经酮基合酶StlD催化肉桂酰辅酶A延伸两个碳单元生成肉桂酰β-酮乙基-StlE(cinnamoyl β-ketoacyl-StlE)。另一合成前体来源于亮氨酸,经支链酮酸脱氢酶BkdA/BkdB催化生成异戊酰辅酶A,然后酮基合酶BkdC催化异戊酰辅酶A与1分子丙二酰辅酶A缩合延伸2个碳单元生成异戊酰-β-酮乙基-ACP。最后,环合酶StlC催化2个硫酯中间体缩合环化生成tapiranof。
基于StlD/StlC从肉桂酰辅酶A连续催化生成tapiranof的功能,Bode课题组[90]对StlD/StlC的同源蛋白DarB/DarA[来源于松噬几丁质菌(Chitinophagapinensis),氨基酸序列一致性大于40%]进行了体外反应和异源表达研究,发现DarB可催化两个β-酮基底物发生Claisen缩合(第一个碳碳键形成)和Michael加成反应(第二个碳碳键形成),生成4-羧基-2,5-二烷基环己烷-1,3-二酮(4-carboxy-2,5-dialkylcyclohexane-1,3-dione),并伴随有脱羧产物2,5-二烷基环己烷-1,3-二酮的产生,随后芳香化酶DarA能够将两种产物均转化为2,5-二烷基间苯二酚(2,5-dialkylresorcinol)化合物。研究人员从DarB/DarA的催化机制推测出,StlD/StlC可能也是利用相同的催化机制形成tapiranof。2016年Abe课题组[91]首次对StlD/StlC进行了体外功能表征,证实了StlD/StlC可分别催化缩合反应和芳构化反应生成二苯乙烯产物。同时,研究人员获得了StlD的X射线晶体结构,氨基酸突变分析表明在保守的催化三联体Cys-His-Asn中His302并不是酶反应的必需氨基酸,而是Glu154作为碱基催化剂激活β-酮基中间体结合到催化位点Cys126上。此外,在结构中也发现由Glu154和Ser340形成的氢键网络激活亲核水分子的存在,表明StlD是利用一种新颖的酶催化机制进行两个酰基底物的缩合形成环己烷二酮骨架。首先,β-酮酰基部分装载到催化残基Cys126上,旋转到短酰基结合口袋。碱性残基Glu154从β-酮酰基单元的γ-碳上提取一个质子,然后在β-酮酰基单元和第二个α,β-不饱和酰基底物之间发生Claisen缩合反应生成支链中间体。然后,酮-烯醇互变异构反应重新激活催化残基Glu154。β-酮酰基部分通过Michael加成反应连接到α,β-不饱和酰基单元的β-碳上产生2,5-二烷基环己烷-1,3-二酮骨架。被激活的水分子发生亲核进攻反应使酶结合中间体的硫酯键断裂。最后,酮-烯醇互变异构反应重新激活催化残基Glu154进入下一反应循环(图8)。
图8
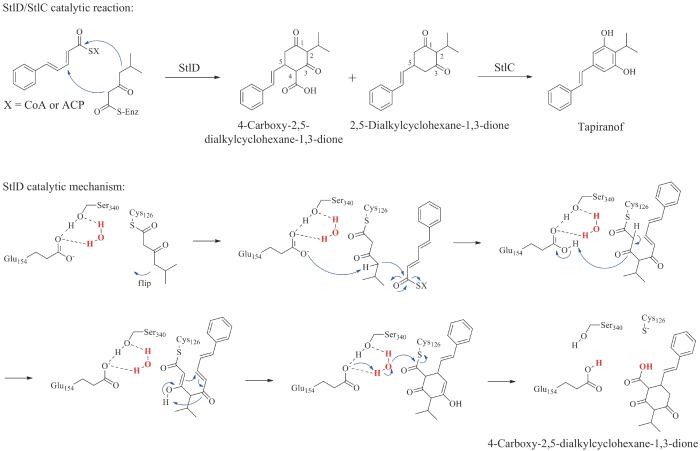
图8 StlD/StlC催化形成tapiranof的反应及StlD的酶催化机制
2022年Bode课题组[92]又对tapiranof的生物合成途径中StlD催化缩合反应之前的5-苯基-2,4-戊二烯酰-StlE(5-phenyl-2,4-pentadienoyl-StlE)中间体的合成进行了进一步的研究。通过体外酶反应发现StlB可替代“缺失的”酰基转移酶能对底物肉桂酸进行活化以及StlE酰基化形成酶结合中间体肉桂酰-StlE,而肉桂酰辅酶A的碳链延伸则是通过Ⅱ型脂肪酸合酶的生物合成得以实现。因此,tapiranof的生物合成是初级代谢途径与特殊代谢途径的交叉结合。此外,Crawford课题组[93]发现在百草枯胁迫下,发光杆菌可转化tapinarof成为具有新颖二聚体结构的药物代谢产物,对炎症细菌具有抑制作用,从结构和功能上拓展了与炎症性疾病相关的抗生素的种类。
3.2 大麻二酚
大麻二酚(cannabidiol,CBD)是2018年FDA批准的药物Epidiolex的主要活性成分,用于治疗与伦诺克斯-加斯托(Lennox-Gastaut)综合征、Dravet综合征或结节性硬化症相关的癫痫发作。大麻二酚在镇痛、抗惊厥、肌肉松弛、抗焦虑、抗精神病方面表现出了良好的效果,可以缓解中度至重度神经性疼痛或其他疼痛状况(如癌症),同时显示出神经保护、抗炎和抗氧化活性[94-95]。大麻二酚是大麻植物(Cannabissativa L.)树脂中天然存在的一种大麻素(cannabinoids),是大麻属植物中发现的至少85个活性大麻素成分[如大麻酚(cannabinol)、大麻萜酚(cannabigerol)、次大麻二酚(cannabidivarin)、Δ9-四氢大麻素(Δ9-tetrahydrocannabivarin)等]之一[96]。大麻二酚是在大麻雌花的腺毛或“大麻芽”中合成的,没有毛状体的雄花通常含量很低,其是作为植物保护剂合成,通常以酸性的大麻二酚酸(cannabidiolic acid,CBDA)形式存在,在储存、加热或碱性条件下,脱羧形成更为常见的大麻二酚形式[97-98]。
大麻二酚的生物合成起始于Ⅲ型聚酮合酶TKS(tetraketide synthase)催化1分子起始单元己酰辅酶A(hexanoyl-CoA)与3分子延伸单元丙二酰辅酶A连续缩合生成3,5,7-三氧代十二烷酰辅酶A(3,5,7-trioxododecaneoyl-CoA)。最初认为3,5,7-三氧代十二烷酰辅酶A可继续发生自发的环化和芳构化反应直接形成橄榄酸(olivetolic acid),然而TKS的体外酶反应仅得到橄榄酸的脱羧形式橄榄醇(olivetol),因此橄榄酸的形成可能不仅仅依赖于单独的TKS,随后橄榄酸环化酶(olivetolic acid cyclase,OAC)的发现证实了这一猜测,3,5,7-三氧代十二烷酰辅酶A经OAC催化发生环合、芳构化以及水解产生橄榄酸,在没有OAC存在的条件下TKS形成的聚酮中间体会发生非酶催化的C2→C7醛缩合并脱羧反应形成橄榄醇[98-101]。虽然橄榄酸的形成依赖于TKS和OAC共同发挥作用,但发现二者之间并不存在直接的相互作用,因此形成的中间体可能是从TKS经细胞溶质转移到OAC。2020年Scrutton课题组[101]获得了TKS与CoA的复合物晶体结构(PDB ID:6GW3),通过突变分析证实了区分Ⅲ型聚酮合酶中环化途径的潜在残基,否定了在此之前提出的存在“醛醇开关”序列释放线性聚酮中间体的猜想,提出了Ⅲ型聚酮合酶催化环化机制的活性位点水活化模式。早在2016年Morita课题组[102]获得了OAC的apo结构以及OAC与橄榄酸的复合物晶体结构。晶体结构分析发现OAC拥有一个特殊的活性空腔,包含疏水性的戊基结合口袋和聚酮结合位点,突变分析发现OAC的Tyr72和His78分别与橄榄酸形成氢键和π-π相互作用并作为酸碱催化剂辅助环化反应(图9)。
图9
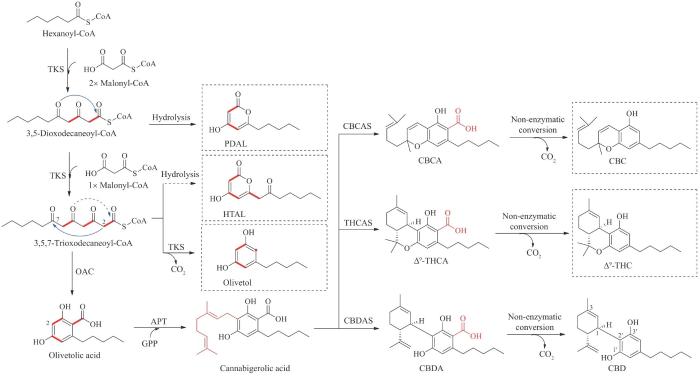
图9 大麻二酚及其他大麻素类似物的生物合成
APT—芳香异戊烯基转移酶;CBDAS—大麻二酚酸合酶;THCAS—四氢大麻酚酸合酶;CBCAS—大麻色烯酸合酶;Δ9-THCA—Δ9-四氢大麻酚酸;CBCA—大麻色烯酸;Δ9-THC—Δ9-四氢大麻酚;CBC—大麻色原烯;PDAL—戊二乙酸内酯;HTAL—己酰三乙酸内酯;GPP—牻牛儿基焦磷酸
TKS和OAC连续催化形成橄榄酸后,芳香异戊烯基转移酶在橄榄酸间苯二酚骨架上高度亲核的C2位插入异戊烯基,生成核心中间产物大麻萜酚酸(cannabigerolic acid,CBGA),最终以CBGA为分支点进一步生成CBDA、大麻二酚及其他大麻素类衍生物(如Δ9-THC、CBC)[98-99,101,103-104]。其中将CBGA转化为CBDA的CBDAS至今仍未获得其晶体结构,但其与THCAS具有84%的序列一致性,都是具有类似催化机制的黄素酶。在活性位点处通过与His114和Cys176形成两个共价键以固定辅因子FAD,Tyr484和His292分别以氢键和静电作用固定底物CBGA,以促进氢化物转移到FAD,通过碳正离子机制发生环化反应,产生具有特定立体构型的环化产物。然而,CBDAS与THCAS在质子转移的催化机制上又略有不同,THCAS从CBGA的羟基上提取一个质子,而CBDAS在CBGA的末端甲基上提取一个质子,这个区域选择性上的差别导致了二者在环化产物上的不同(图10)[26,105]。环化产物CBDA很容易通过非酶热脱羧反应获得中性大麻素大麻二酚,虽然大麻二酚在植物中浓度不显著,但近些年通过气相及液相色谱方法对酸性及中性大麻素的实时测定,推测与邻酚形成分子内氢键可能是其热脱羧机制[100,106-107]。
图10
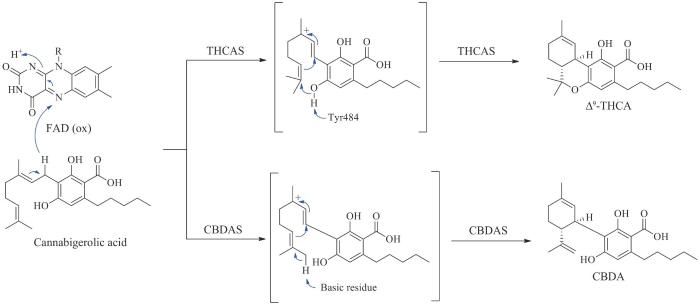
图10 CBGA转化为CBDA和Δ9-THCA的不同的生物合成机制
但是在大麻素类成分的研究与开发中一直受到很多因素的限制,例如在植物中的丰度较低、结构复杂难以大规模合成以及法律方面的因素等。2019年Keasling课题组[108]在大麻素异源合成方面实现了重大突破,通过对酿酒酵母的工程化改造,引入己酰辅酶A合成途径、橄榄酸合成基因、牻牛儿基转移酶以及大麻素合酶基因,成功合成了CBGA、Δ9-THCA、CBDA、次大麻二酚酸(cannabidivarinic acid,CBDVA)以及Δ9-四氢次大麻酚酸(Δ9-tetrahydrocannabidivarinic acid,THCVA)等多个大麻素成分。2020年Bowie课题组[109]又利用组合酶催化的方法实现了大麻素CBGA的生物合成,产量相比在酵母中的异源合成有了进一步的提高,达到了0.5 g/L(图11)。总体来说,酵母异源合成系统与体外无细胞催化系统的建立,不仅可以实现大麻素类活性成分的大规模合成,同时也为更多新颖活性的天然和非天然大麻素的挖掘及创新提供了新的途径。
图11
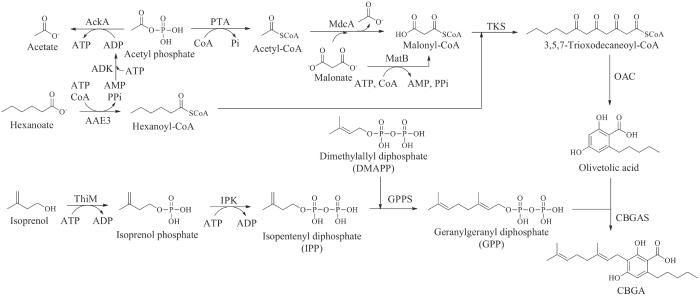
图11 CBGA在体外的酶法生物合成
AckA—乙酸激酶;PTA—磷酸乙酰基转移酶;MdcA—丙二酸脱羧酶;MatB—丙二酰辅酶A合成酶;ADK—腺苷酸激酶;AAE3—酰基激活酶;ThiM—羟乙基噻唑激酶;IPK—磷酸激酶;GPPS—牻牛儿基焦磷酸合酶
3.3 奥利司他
奥利司他(orlistat)是一款FDA在1999年批准的老药,用于治疗肥胖症。奥利司他通过可逆抑制胃和胰腺脂肪酶而起作用,脂肪酶的失活阻止了甘油三酯的水解,使游离脂肪酸在体内不被吸收,同时奥利司他也具有心血管安全性和控制肥胖患者糖尿病的益处。此外,由于奥利司他能够抑制肿瘤细胞脂肪酸合酶的硫酯酶结构域,因此还具有一定的抗肿瘤活性[110-112]。
奥利司他是利普司他汀(lipstatin)的烷基链饱和衍生物。利普司他汀是一种有效的天然胰腺脂肪酶抑制剂,于1987年从毒三素链霉菌(Streptomycestoxytricini)中首次分离得到[113]。利普司他汀是一个2,3-反式二取代的β-内酯结构,两个取代基均是线性的烷基链,分别为在α位的C6链和在β位的C13链,此外,还有一个N-甲酰-L-亮氨酸残基以酯键形式连接于β链C5位的羟基上。β-内酯环是发挥脂肪酶抑制作用的关键结构,β-内酯环的开环可导致利普司他汀的活性完全丧失。自从利普司他汀被发现以来,其生物合成机制一直进展缓慢,仅通过同位素标记实验对利普司他汀的前体来源进行了研究,发现利普司他汀的α支链脂肪酸基本骨架是由辛酰辅酶A(octanoyl-CoA)和3-羟基十四烷-5,8-二烯酰辅酶A(3-hydroxytetradeca-5,8-dienoyl-CoA)通过Claisen缩合反应得到,二者均来自于亚油酸(linoleic acid)的β-氧化产物[114-118]。
直到近十年,利普司他汀的生物合成才有了突破性进展。2014年,陶美凤课题组[119]首次在毒三素链霉菌中鉴定了利普司他汀的生物合成操纵子lst,lst共包含6个基因lstA~F,根据体内基因敲除实验结果,研究人员推测了利普司他汀完整的生物合成过程。首先,酰基辅酶A合成酶(acyl-CoA synthetase)LstC催化ATP依赖的辛酸(octanoic acid)活化及CoA连接产生辛酰辅酶A,辛酰辅酶A经一个未知的酰基辅酶A羧化酶(acyl-CoA carboxylase,ACCase)复合物催化得到己基丙二酰辅酶A(hexylmalonyl-CoA),再经初级代谢途径转移到ACP上形成己基丙二酰-ACP硫酯中间体,作为骨架缩合的底物之一。另外一个底物为C14链的3-羟基十四烷-5,8-二烯酰辅酶A,由亚油酸被活化后产生的亚油酰辅酶A(linoleoyl-CoA)经β氧化得到。两个底物被两个Ⅲ型β-酮酰基-酰基载体蛋白合酶(β-ketoacyl-acyl carrier protein synthases Ⅲ,KAS Ⅲ)LstA和LstB协同催化,发生连续的脱羧反应以及Claisen缩合反应,形成3-酮基-5-羟基-C22-ACP硫酯中间体。另一方面,非核糖体肽合成酶(nonribosomal peptide synthetase,NRPS)LstE活化亮氨酸并上载到肽基载体蛋白(peptidyl carrier protein,PCP)结构域上,甲酰基转移酶(formyltransferase)LstF转移甲酰基到PCP连接的亮氨酸残基的α-氨基上,然后LstE催化C5位的羟基亲核进攻PCP连接的亮氨酸残基的酰基碳,形成酯键连接的N-甲酰-L-亮氨酸-C22-ACP硫酯中间体。最后,3β-羟基类固醇脱氢酶LstD还原C3位酮基为羟基,产生的羟基可能自发亲核进攻ACP连接的中间体的酰基碳,最终形成β内酯结构(图12)。
图12
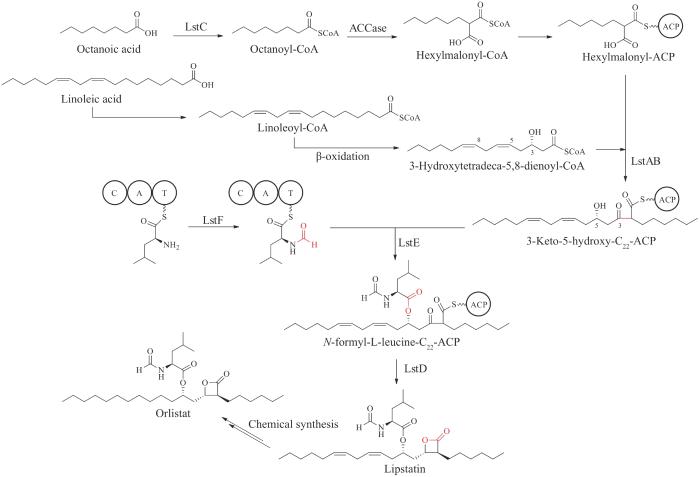
图12 奥利司他化学结构及推测的利普司他汀生物合成途径
C—缩合结构域;A—腺苷化结构域;T—硫酯化结构域
2019年刘文课题组[50]对Ⅲ型KAS LstAB的催化机制进行了进一步的深入研究,发现利普司他汀生物合成中的LstAB是以独特的异源二聚体形式存在,可以催化C8与C14酰基辅酶A发生非脱羧形式的缩合,而常见的KASⅢ(如E.coli来源的FabH)为典型的同源二聚体,通常催化乙酰辅酶A与丙二酰-ACP之间脱羧形式的Claisen缩合。通过LstAB的体外分析发现,形成的缩合产物C22 α-烷基-β-酮酸非常不稳定,会自发脱羧形成C21产物,而LstD可将不稳定的C22 α-烷基-β-酮酸还原为稳定的C22 α-烷基-β-羟基酸。结合LstAB催化残基的鉴定,研究人员提出了LstAB可能的催化机制(图13)。大亚基LstA含有典型的Cys-His-Asn催化三联体,可能与C8酰基辅酶A反应形成酰基-Cys酶中间体。相比之下,小亚基LstB缺乏这种三联体,但具有Glu催化残基,它以底物依赖的方式作用于C8酰基-Cys酶中间体,作为Cα去质子化的碱或作为Michael型加成引发级联反应的亲核试剂,产生烯醇负离子,通过单向亲核取代进行C14酰基-辅酶A头对头的组装。最后利用生物信息学手段,发现lstAB的同源基因广泛分布于各类细菌基因组中,表明LstAB这种以异源二聚体形式催化非脱羧缩合的方式广泛存在于与利普司他汀类似的天然产物的生物合成中。
图13
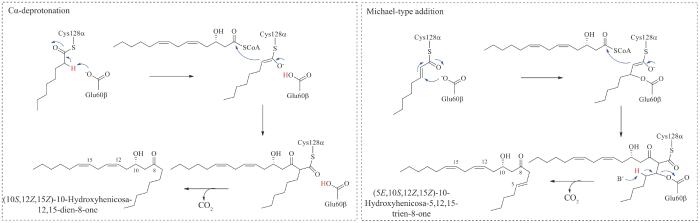
图13 推测的LstAB非脱羧缩合机制
4 非核糖体肽类天然产物
4.1 曲贝替定
曲贝替定(trabectedin)也称为海鞘素743(ecteinascidin 743,ET-743),是第一个海洋来源的新型抗软组织肿瘤药物,于1984年从加勒比海被囊动物红树海蛸(Ecteinascidiaturbinata)体内分离得到。ET-743独特的作用机制来源于它的醇胺部分,可以转化为亲电性的亚胺离子,与DNA的小凹槽结合发生烷基化,干扰细胞分裂、基因转录过程和DNA修复机制,在欧洲、俄罗斯和韩国等多地批准用于治疗晚期软组织肉瘤。2015年,FDA首次批准以ET-743为活性成分的新药Yondelis,用于针对晚期软组织肉瘤和卵巢癌的抗肿瘤药物[120-124]。
长期以来研究人员一直认为红树海蛸可能并不是ET-743的实际产生生物,推测其可能真正来源于一种基因组未知的微生物。因为难以在实验室条件下培养该微生物,因此Sherman课题组[125-126]对从被囊动物中分离得到的宏基因组DNA进行了测序组装,发现ET-743的实际产生生物为一种γ-变形杆菌纲的共生细菌CandidatusEndoecteinascidiafrumentensis,并对ET-743的四氢异喹啉核心组装酶的预测活性进行了体外验证,从而确定了其真实来源。ET-743为四氢异喹啉类天然产物,具有一个特征性的五环结构,与其类似的结构也在多个天然产物中发现,如番红霉素(saframycin)MX1、番红霉素A、番红菌素(safracin)B、萘啶霉素(naphthyridinomycin,NDM)以及喹诺卡因(quinocarcin,QNC)等[127-131],虽然关于ET-743的生物合成研究进展较少,但对这些四氢异喹啉类天然产物的生物合成研究也有助于揭示ET-743的生物合成途径。
目前推测ET-743主要骨架由功能罕见的NRPSs EtuA1-A3催化完成,其生物合成前体为1分子丝氨酸残基、1分子羟乙酰基团以及2分子酪氨酸衍生物3-羟基-5-甲基-氧甲基酪氨酸(3-OH-5-Me-OMe-Tyr)。ET-743的生物合成起始与番红霉素A的生物合成途径类似,NRPS EtuA3的酰基辅酶A连接酶结构域催化激活一个未知的长链脂肪酸上载到ACP上,然后在EtuA3的延伸模块缩合1分子的半胱氨酸残基形成PCP-N-酰基半胱氨酰硫酯(PCP-N-acylcysteinyl thioester)中间体[132]。ET-743的第二个前体单元与NDM和QNC的生物合成类似,来源于5-磷酸戊酮糖(pentulose-5-phosphate),经转酮酶-ACP复合物催化将1分子羟乙酰基单元转移到ACP上形成ACP-羟乙酰硫酯延伸单元[133],然后EtuA1催化PCP-N-酰基半胱氨酰硫酯与ACP-羟乙酰硫酯的缩合延伸形成酰基化酯肽硫酯中间体,经EtuA2的末端还原结构域TR(与番红霉素NRPS TR结构域功能类似)还原释放产生醛中间体Int 1。ET-743的第三个前体单元来源于酪氨酸,依次经过羟基化(EtuH催化)、碳甲基化(EtuM2催化)以及氧甲基化(EtuM1催化)形成衍生物3-OH-5-Me-OMe-Tyr(与番红霉素和番红菌素生物合成类似)[134-135]。EtuA2识别3-OH-5-Me-OMe-Tyr并上载到PCP上,并通过其特殊的缩合结构域与酰基化酯肽硫酯缩合产生亚胺中间体,同时发生Pictet-Spengler反应产生新的四氢吡啶环,随后再经EtuA2 TR结构域还原释放产生第二个酰基化酯肽硫酯中间体Int 2。发生Pictet-Spengler反应的特殊EtuA2缩合结构域含有保守的HxxxxD基序,区别于典型的HHxxxDG基序[132]。
第二个酰基化酯肽硫酯中间体可以继续与一分子的3-OH-5-Me-OMe-Tyr硫酯中间体缩合及Pictet-Spengler反应形成第二个四氢吡啶环,并经第三次EtuA2 TR结构域的还原释放产生第三个酰基化酯肽硫酯中间体Int 3,该中间体可能自发环化,形成半缩醛胺产物中间体Int 4。随后其上的N-酰基脂肪酸链可能由ET-743基因簇中的青霉素酰化酶同源蛋白EtuF3切除产生中间体Int 5,Int 5经连续的氧化及乙酰化(EtuY及EtuO1/2/3/4,EtuO1/2/3/4与番红霉素生物合成途径的SfmO2蛋白同源)得到Int 6,Int 6在一个未知功能酶的作用下发生环化反应形成硫醚中间体ET-583[136]。随后N-甲基转移酶EtuM4催化ET-583的二级胺发生甲基化产生ET-597,ET-597在另一个未鉴定酶的催化下发生氧化脱氨反应得到ET-596,FAD依赖的单加氧酶EtuO1/2/3/4催化ET-596形成亚甲基二氧桥环产物ET-594,最后EtuA2缩合结构域催化ET-594与3-OH-OMe-Tyr硫酯中间体发生第三次缩合及Pictet-Spengler反应,最后经还原释放得到最终的产物ET-743(图14)[29,137]。
图14
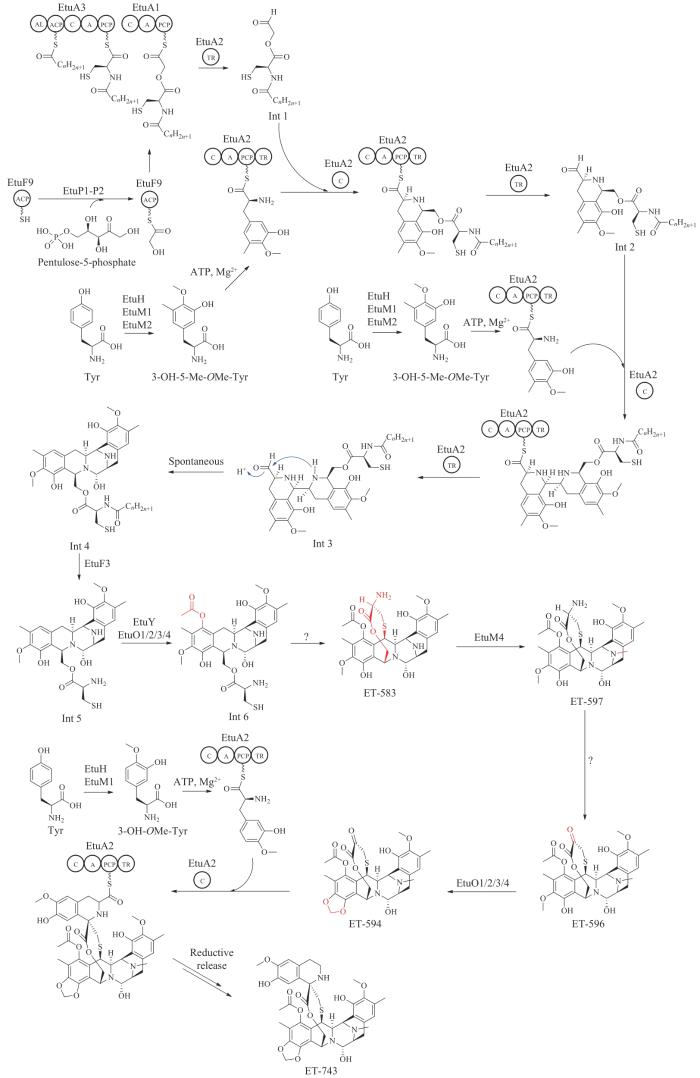
图14 推测的ET-743生物合成途径
AL—酰基辅酶A连接酶结构域;TR—末端还原酶结构域
4.2 瑞扎芬净
瑞扎芬净(rezafungin)是半合成棘白菌素(echinocandin)类药物。瑞扎芬净能抑制真菌细胞壁中的1,3-β-D-葡聚糖合酶复合物,是一种有效的抗真菌药物,具有较高的特异性和较好的耐受性,对白色念珠菌(Candidaalbicans)、光滑念珠菌(Candidaglabrata)、近平滑念珠菌(Candidaparapsilosis)、热带念珠菌(Candidatropicalis)以及其他多种念珠菌和曲霉菌具有抗菌活性。2023年FDA批准了以瑞扎芬净为活性成分的药物Rezzayo,用于治疗成人念珠菌病和侵袭性念珠菌病。此外,Rezzayo目前也正在研究用于预防接受同种异体血液和骨髓移植的成人侵袭性真菌疾病[138-139]。
瑞扎芬净的半合成前体棘白菌素是药物研究和开发的杰出范例。棘白菌素类天然产物是由6个氨基酸缩合而成的环脂肽分子结构,其中5个氨基酸为非蛋白原氨基酸,形成一个非常保守的六肽骨架。最早的棘白菌素类天然产物——棘白菌素B于1974年在一种曲霉菌(Aspergillusdelacroxii,原名为Aspergillusnidulans var.echinolatus A 32204)的代谢物中首次发现[140],截至目前,已在超过20种真菌中发现了棘白菌素B及其类似物,其中棘白菌素B是最为常见的代谢产物,主要分布在曲霉属和裸胞壳属(Emericella)真菌中[141-146]。由于棘白菌素复杂的结构难以进行化学全合成,为了提高该类天然产物的活性和溶解度,化学半合成一直是将该类化合物发展为潜在药物的有效手段,截至目前FDA批准的半合成棘白菌素类药物除瑞扎芬净以外,还有卡泊芬净(caspofungin,2001年批准)、米卡芬净(micafungin,2005年批准)以及阿尼芬净(anidulafungin,2006年批准)[147-149]。
对棘白菌素类天然产物的生物合成研究在2012年才取得重要突变,唐奕课题组[150]在一株曲霉菌(Aspergilluspachycristatus,原名为Emericellarugulosa NRRL 11440)中发现并表征了棘白菌素B的生物合成基因簇。棘白菌素B的基因簇最初认为是由基因组上两个不同的基因簇(ecd和hty)组成,之后通过序列比较和PCR实验表明,它实际上是一个连贯的生物合成基因簇(ecd/hty)[151]。棘白菌素B的生物合成基因簇包含1个六模块的NRPS EcdA、1个酰基-AMP(acyl-AMP)连接酶EcdI、5个加氧酶EcdGHK和HtyEF以及负责L-高酪氨酸(L-homotyrosine)合成的4个酶HtyABCD。L-高酪氨酸的合成途径推测为:首先4-羟基苯基丙酮酸在异丙基苹果酸合酶HtyA的催化下与乙酰辅酶A发生醛缩合反应,释放CoA的同时生成2-(4-羟基苯基)苹果酸,2-(4-羟基苯基)苹果酸经顺乌头酸酶(aconitase)HtyD的异构化作用转化为3-(4-羟基苯基)-苹果酸;随后,异丙基苹果酸脱氢酶HtyC催化3-(4-羟基苯基)-苹果酸发生脱羧及氧化反应产生4-(4-羟基苯基)-2-氧代丁酸,最后经转氨酶HtyB催化形成L-高酪氨酸(图15)。
图15

图15 推测的棘白菌素B生物合成途径
棘白菌素B的生物合成起始于酰基-AMP连接酶EcdI活化亚油酸生成亚油酰-AMP,然后转移到NRPS EcdA的硫酯化结构域T0,随后经6个EcdA延伸模块缩合形成线性六肽,线性六肽在EcdA的末端缩合结构域CT的催化下发生大环化,释放环形骨架中间体,最后经多次羟基化修饰得到最终的产物棘白菌素B(图15)。关于羟基化修饰在合成途径中发生的位置,Walsh课题组[152]对加氧酶EcdGHK进行了基因敲除及体外酶活研究。研究发现,细胞色素P450加氧酶EcdH催化的加氧反应发生在环形骨架(推测为中间体Int 1)形成之后,在鸟氨酸残基上形成邻二醇结构,而Fe2+/α-酮戊二酸(Fe2+/α-ketoglutarate,α-KG)依赖的加氧酶EcdG仅催化L-高酪氨酸的C3位发生羟基化,另一个Fe2+/α-KG依赖的加氧酶EcdK实际为亮氨酸5-羟化酶,催化亮氨酸C5位发生氧化产生γ-Me-Glu-γ-半醛与环亚胺的平衡产物,环亚胺通过进一步的未鉴定反应形成4R-甲基-L-脯氨酸进入棘白菌素B的生物合成途径。2018年Hüttel课题组[153]证实第6位脯氨酸的C3位羟基化由Fe2+/α-KG依赖的加氧酶HtyE催化。近些年,通过对该家族的其他棘白菌素类天然产物(包括Pneumocandin B0和FR901379)的生物合成研究,确定了另外两个羟基化修饰由细胞色素P450加氧酶HtyF催化产生[154-156]。
截至目前,已对约10个棘白菌素类天然产物的生物合成基因簇进行了研究,系统发育分析表明,棘白菌素类天然产物的生物合成是从共同的祖先进化而来,在2.9亿到3.9亿年前分化到两个不同的真菌分支(锤舌菌纲和散囊菌纲)[157-159]。最近,在沙地青霉(Penicilliumarenicola NRRL 8095)中也发现了杂合的生物合成基因簇,推测为棘白菌素生物合成基因簇水平基因转移的结果[160-161]。由于该家族的天然产物是重要的抗真菌药物来源,近些年对棘白菌素B类似物的生物合成以及产生菌的工程化改造方面都取得了很大的进展[162-164]。
5 木质素类天然产物——依托泊苷
依托泊苷(etoposide)是FDA在1983年批准的化疗药物,可以治疗多种不同类型的癌症(如睾丸和小细胞肺肿瘤)。依托泊苷与拓扑异构酶Ⅱ和DNA形成复合物,该复合物诱导DNA双链断裂,同时阻止拓扑异构酶Ⅱ结合修复,达到抑制DNA合成并导致细胞死亡的作用[165]。
依托泊苷是鬼臼毒素(podophyllotoxin)的半合成衍生物。鬼臼毒素属于木脂素类天然产物,从药用植物桃儿七(Podophyllumhexandrum)中分离得到[166]。目前,鬼臼毒素的生物合成仅有部分已知,主要涉及从松柏醇(coniferyl alcohol)到(-)-pluviatolide的鬼叶毒素早期生物合成途径[167-170]。2015年,Sattely课题组[171]通过转录组挖掘以及组合生物合成手段,从桃儿七中鉴定了六个新的途径酶,包括一个2-酮戊二酸依赖的双加氧酶,负责催化新的碳碳键形成,关闭aryltetralin支架的核心环己烷环(图16)。将六个新的途径基因与四个已知的基因在烟草中共表达后,获得了依托泊苷苷元前体(-)-4′-desmethylepipodophyllotoxin。该研究结果实现了依托泊苷苷元在烟草中的异源生产,相比于在化学半合成中需要差向异构化和去甲基化,依托泊苷苷元的异源合成更为简单高效。2019年Sattely课题组[172]又利用已经鉴定的生物合成(-)-deoxypodophyllotoxin的8个基因,结合合成前体松柏醇的8个基因,共计16个基因在异源宿主烟草中实现了共表达,在烟草叶子中累积的产量达到4.3 mg/g,使依托泊苷前体的生物全合成提升到了毫克级别。
图16
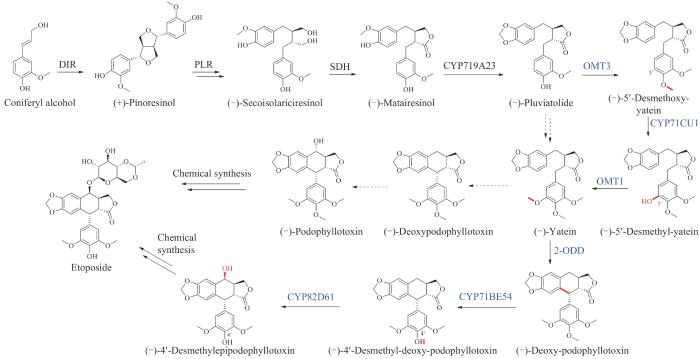
图16 依托泊苷苷元的生物合成途径
(蓝色为新表征的酶)DIR—定向蛋白;PLR—松脂醇-落叶松脂醇还原酶;SDH—开环异落叶松脂酚脱氢酶;CYP719A23/71CU1/71BE54/82D61—细胞色素P450;OMT1/3—O-甲基转移酶;2-ODD—2-酮戊二酸/Fe(Ⅱ)依赖双加氧酶
6 生物碱类天然产物
6.1 长春花碱和长春新碱
长春花碱(vinblastine)是20世纪60年代FDA批准的药物,是一种有效的抗癌药物,它可以结合微管蛋白,从而抑制微管的组装。长春花碱通常与其他药物一起使用,用于治疗多种类型的癌症,包括霍奇金淋巴瘤、非小细胞肺癌、膀胱癌、脑癌、黑色素瘤和睾丸癌。长春新碱(vincristine)也是在同时期批准的药物,用于治疗急性白血病、恶性淋巴瘤和急性全髓增殖症等[173-175]。
长春花碱和长春新碱均是两个单萜吲哚生物碱的异二聚体,最早于1958年从马达加斯加长春花(Catharanthusroseus)中分离得到。长春花碱及其前体的生物合成途径仍然有部分未知,尤其是从前体stemmadenine合成中间体长春质碱(catharanthine)和水甘草碱(tabersonine)的催化酶仍然未鉴定[176-177]。2018年O’Connor课题组[178]利用转录组分析技术鉴定了长春花碱生物合成途径中两个缺失的氧化还原酶precondylocarpine acetate synthase(PAS)和dihydroprecondylocarpine acetate synthase(DPAS)。研究发现,PAS和DPAS可将stemmadenine acetate转化为一种不稳定的dihydroprecondylocarpine acetate,然后被两种水解酶长春质碱合酶(catharanthine synthase,CS)或水甘草碱合酶(tabersonine synthase,TS)中的一种去乙酰化,通过Diels-Alder环化生成长春质碱或水甘草碱,最终二聚生成长春花碱和长春新碱。在异源宿主烟草中共表达PAS、DPAS以及CS或TS,并喂养stemmadenine acetate后成功实现了长春质碱或水甘草碱的生物合成,为半合成制备长春花碱或长春新碱提供了化学原料(图17)。2022年Keasling课题组[179]将长达30个酶促步骤的长春花碱生物合成途径重构在高度工程化的酵母细胞中,从头合成了长春花碱的前体文多林(vindoline)和长春质碱,并在体外通过化学偶联的方法实现了长春花碱的全合成。2023年连佳长课题组[180]利用底盘菌株毕赤酵母以简单的碳源从头合成了长春质碱,合成滴度高达2.57 mg/L。利用酵母细胞等可以生产复杂植物天然产物的优势工厂,有望在不久的将来实现具有高附加值的长春花碱的生物全合成。
图17
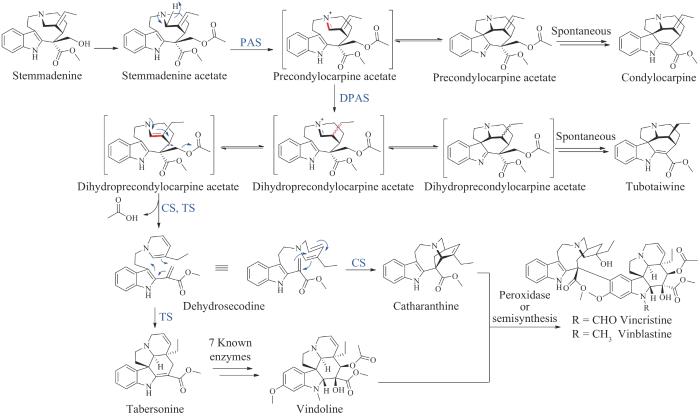
图17 长春花碱和长春新碱的生物合成途径
(蓝色为新表征的酶)
6.2 秋水仙碱
秋水仙碱(colchicine)是FDA在1961年首次批准的药物,主要用于治疗痛风、白塞病和家族性地中海热病以及预防心肌梗死等[181-183]。秋水仙碱可以与微管蛋白二聚体相互作用从而抑制微管生长,但对微管动力学的影响与减轻炎症之间的关系目前仍不完全清楚[184-185]。
秋水仙碱来源于秋水仙属(Colchicum)和嘉兰属(Gloriosa)植物,是具有三环结构的生物碱,包括国内课题组在内的研究学者在秋水仙碱的化学合成方面已经取得了不少进展,可达到克级规模,但秋水仙碱在植物中的生物合成途径一直是一个未解之谜[186-187]。直到2020年Sattely课题组[53]首次解析了秋水仙碱近乎完整的生物合成途径。基于前期在秋水仙属植物中大量的同位素标记实验结果,以及与秋水仙碱相关的秋水仙科生物碱的结构表征,研究人员推断秋水仙碱的核心骨架来源于L-苯丙氨酸和L-酪氨酸[188-189]。然后,通过转录组挖掘手段,从植物嘉兰(Gloriosasuperba)中发现了生物合成秋水仙碱前体N-formyldemecolcine的8个基因,推测秋水仙碱的生物合成首先从L-苯丙氨酸和L-酪氨酸分别合成前体4-羟基二氢肉桂醛(4-hydroxydihydrocinnamaldehyde,4-HDCA)和多巴胺,由Pictet-Spengler反应缩合生成含有1-苯基异喹啉骨架的中间体,经过连续多步甲基化、羟基化反应后生成中间产物(S)-autumnaline,(S)- autumnaline经苯酚对位碳-碳偶联和甲基化反应生成中间产物O-methylandrocymbine,最后通过细胞色素P450酶CYP71FB1催化的氧化扩环反应形成具有环庚三烯酚酮结构的秋水仙碱前体N-formyldemecolcine。此外,研究人员还通过代谢工程手段在本氏烟草中重构了N-formyldemecolcine的生物合成途径,实现了从简单氨基酸前体到具有独特碳骨架的秋水仙碱类生物碱的生物全合成,为更多类似药用植物天然产物生物合成途径的发现和工程化打下基础(图18)。
图18
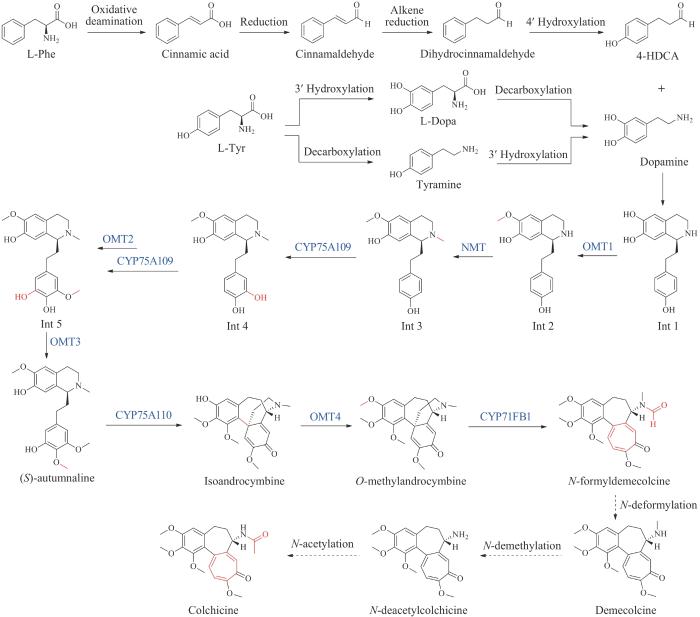
图18 秋水仙碱的生物合成途径
(蓝色为新表征的酶)OMT1/2/3/4—O-甲基转移酶;NMT—N-甲基转移酶;CYP75A109/75A110/71FB1—细胞色素P450
6.3 吗啡和可待因
吗啡(morphine)和可待因(codeine)是FDA批准的天然阿片类药物。吗啡最初于1941年批准使用,用于缓解中度至重度急性和慢性疼痛[190]。可待因于1950年获得批准,是一种通过提高疼痛阈值而不损害意识或改变其他感觉功能来减轻疼痛的药物,可用作中枢镇痛药、镇静药、安眠药、抗神经病变性疼痛药和抗肠蠕动药,也用于某些持续咳嗽的疾病[191]。
吗啡和可待因均是来源于植物罂粟(Papaversomniferum)的苯基异喹啉生物碱,在过去的30年里,吗啡和可待因的生物合成途径几乎已经完全解析,除了少数步骤仍不明确,其中包括(S)-牛心果碱(reticuline)差向异构化为(R)-牛心果碱的关卡步骤。2015年Graham课题组[192]发现催化这一关键步骤的是一个细胞色素P450酶和氧化还原酶双模块融合酶STORR[(S)- to (R)- reticuline]。STORR的细胞色素P450酶模块负责将(S)-牛心果碱氧化为1,2-脱氢牛心果碱,而氧化还原酶模块则负责将1,2-脱氢牛心果碱还原为(R)-牛心果碱。STORR基因的发现具有重要的意义,填补了吗啡烷类生物碱在生物合成中缺失的关键部分(图19)。2019年Facchini课题组[54]又报道了阿片类生物碱生物合成过程中一个新的异构酶NISO(neopinone isomerase)的发现,在此之前由吗啡烷类生物碱的前体neopinone异构化为可待因酮(codeinone)的关键步骤一直认为是自发过程。在工程化的酵母菌株中加入NISO后,显著提高了阿片类药物可待因和氢可酮的产生(图19)。在关键性生物合成步骤上的突破性发现,为今后吗啡烷类生物碱的优化生产提供了更多借鉴。
图19

图19 吗啡和可待因的生物合成途径
(蓝色为新表征的酶)
6.4 东莨菪碱
东莨菪碱(scopolamine)是从茄科(Solanaceae)植物中分离出来的一种托品烷生物碱,于1979年首次获得FDA批准,具有抗胆碱作用,用于治疗与晕动症和术后相关的恶心呕吐[193-194]。东莨菪碱的托品烷基本骨架部分由吡咯环和哌啶环骈合而成。过往的同位素标记研究表明,托品烷基本骨架部分由来源于鸟氨酸的N-甲基吡咯啉阳离子和2分子的乙酰单元形成,但催化其缩合的酶一直未见报道。2019年黄胜雄课题组[195]利用种内转录组差异表达和种间同源基因分析,发现了罕见的Ⅲ型聚酮合酶PYKS,催化N-甲基吡咯啉阳离子与丙二酰辅酶A的缩合,形成托品烷基本骨架合成的关键中间体4-(1-甲基-2-吡咯烷基)-3-氧代丁酸[4-(1-methyl-2-pyrrolidinyl)-3-oxobutanoic acid],并通过蛋白晶体结构解析阐明了该缩合反应的酶学机制,发现PYKS催化2分子丙二酰辅酶A合成三羰基戊二酸中间体,三羰基戊二酸中间体与N-甲基吡咯啉阳离子自发地发生Mannich缩合反应(图20)。2019年廖志华课题组[196]分离鉴定了托品碱生物合成途径中负责托品烷部分与苯乳酸部分酯化连接的两个全新功能的酶,苯乳酸UDP-糖基转移酶UGT1(phenyllactate UDP-glycosyltransferase)和海螺碱合成酶LS(littorine synthase),解决了托品碱的酯键形成机制这一长期难题。此外,该课题组[197-199]还发现了在莨菪碱生物合成途径中其他三个关键酶,包括苯丙酮酸还原酶PPAR(phenylpyruvic acid reductase)、鸟氨酸脱羧酶ODC(ornithine decarboxylase)以及莨菪碱脱氢酶HDH(hyoscyamine dehydrogenase),为东莨菪碱生物合成途径的完整解析做出了重要贡献,同时也为东莨菪碱的细胞生物全合成提供了必需基因(图20)。2019年,Smolke课题组[200]利用简单的碳源和氮源在酿酒酵母中实现了东莨菪碱关键前体托品碱的生物合成,合成滴度达到6 mg/L。2020年,该课题组[201]又同样利用酵母细胞,结合功能基因组学鉴定了途径中缺失的酶,并从简单的糖源和氨基酸开始,实现了药用生物碱东莨菪碱的生物全合成。该合成平台可以促进托品烷生物碱衍生物的发现,有助于将来实现这些基本药物的稳健和灵活供应。
图20
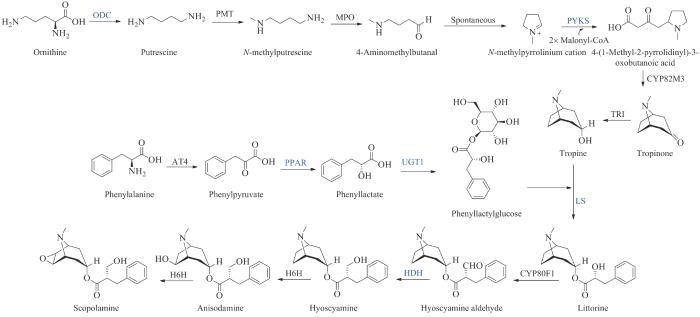
图20 东莨菪碱的生物合成途径
(蓝色为新表征的酶)
7 萜类天然产物——来法莫林
来法莫林(lefamulin)是截短侧耳素(pleuromutilin)类半合成药物,与2007年批准的药物瑞他莫林(retapamulin)具有相同的前体来源。来法莫林通过与细菌50S核糖体亚基的肽基转移酶中心结合,从而阻断细菌蛋白质的合成。2019年FDA首次批准了以来法莫林为活性成分的抗生素药物Xenleta,用于治疗成人细菌性肺炎。来法莫林作为一种新型的截短侧耳素抗生素,对多种革兰氏阳性和革兰氏阴性细菌引起的呼吸道感染具有良好的疗效[202-204]。
来法莫林的半合成前体截短侧耳素属于二萜类天然产物,最早于1951年在担子菌纲侧耳属(Pleurotusmutilu和Pleurotuspasseckerianus,别名Clitopiluspasseckerianus)中分离得到。截短侧耳素的化学结构在1968年时已完全解析,它的主要特征是类似螺桨烷的三环骨架,由五元、六元和八元环组成。同时,研究人员也解析了截短侧耳素三环骨架的环化机制,发现在双(牻牛儿基)焦磷酸盐(geranylgeranyl diphosphate,GGPP)的质子依赖环化过程中,萜类合酶的Ⅱ类结构域催化发生环收缩形成5/6二环骨架。然后,Ⅰ类萜烯合酶结构域通过去磷酸化催化八元环的形成,同时捕获1分子水,在三环骨架的C14位上引入第一个羟基[205-207]。2007年,Oikawa课题组[208]通过氘标记的前体喂养实验,进一步确认了截短侧耳素的生物合成途径中可能的中间体形式。
2016年Foster课题组[209]在侧耳菌(C.passeckerianus)中首次鉴定了截短侧耳素的生物合成基因簇ple,基因簇ple由七个基因组成,分别编码一个GGPP合酶(ple4)、一个萜类合酶(ple3)、三个细胞色素P450酶(ple1/5/6)、一个短链脱氢酶/还原酶(ple7)以及一个酰基转移酶(ple2)。但是,在产生菌C.passeckerianus中难以提高截短侧耳素的产量,因此研究人员在子囊菌异源宿主米曲霉中进行了表达,成功实现了截短侧耳素的异源生物合成,同时产量提高了2106%。2017年该课题组[210]在前期研究基础上,将截短侧耳素的生物合成基因在米曲霉中逐步表达,最终揭示了截短侧耳素完整的生物合成途径。首先,由Ple4合成的GGPP在萜类合酶Ple3催化下发生两次环化形成5/6/8三环中间体premutilin,然后由细胞色素P450酶Ple5和Ple6分别在C11位和C3位发生立体特异性的羟基化反应产生3,11-dihydroxypremutilin,该三元醇中间体被短链脱氢酶Ple7进一步氧化,将C3位的羟基氧化为酮基得到中间产物mutilin,最后经Ple2催化的乙酰基转移反应和Ple1催化的羟基化反应产生最终的产物截短侧耳素(图21)。同年,Oikawa课题组[211]也报道了截短侧耳素生物合成基因在米曲霉中的逐步表达,并将萜类合酶Ple3在体外进行了功能表征,获得了相同的截短侧耳素生物合成解析结果。近年来,利用异源宿主米曲霉、酿酒酵母以及组合生物合成的方法,研究人员又获得了更多新的具有抗菌活性的截短侧耳素类似物,为该家族的药物拓展提供了更多选择[212-214]。
图21
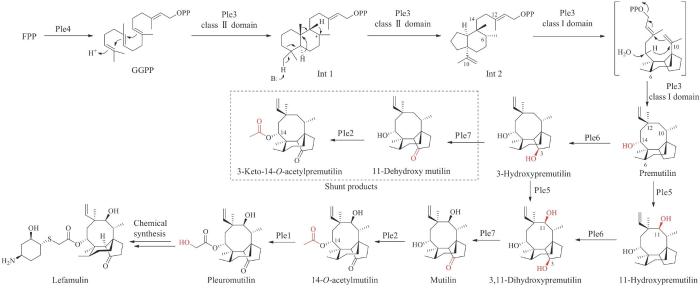
图21 二萜类天然产物截短侧耳素的生物合成途径
8 肽类天然产物——血管紧张素Ⅱ
血管紧张素(angiotensin Ⅱ)是一种导致血管收缩和血压升高的肽激素,血管紧张素也能刺激肾上腺皮质醛固酮的释放,促进肾脏钠贮留[215]。2017年FDA批准了以血管紧张素Ⅱ为主要活性成分的药物Giapreza注射剂,其作为血管收缩剂用于提高患有脓毒性休克或其他分布性休克的成年人血压。该药物是第一个也是唯一一个使用人造血管紧张素Ⅱ来帮助维持血压的药物。
血管紧张素Ⅱ来源于前体分子血管紧张素原(angiotensinogen)。血管紧张素原是一种在肝脏中产生的血清球蛋白,经肾素催化产生血管紧张素Ⅰ(angiotensinⅠ,十肽),血管紧张素Ⅰ含有肾素-血管紧张素系统作用所必需的肽序列,它没有直接的生物活性,仅作为血管紧张素Ⅱ的前体存在。血管紧张素Ⅰ通过血管紧张素转换酶(angiotensin-converting enzyme,ACE)去除两个C末端残基转化为血管紧张素Ⅱ,实现血管紧张素Ⅱ在体内的生物合成(图22)[27,216-217]。
图22

图22 血管紧张素Ⅱ在人体内的生物合成
9 其他天然产物
除了以上FDA批准的天然产物药物,还有一部分更早批准的药物也在近些年获得了较大的研究进展,受到了药物研究领域的广泛关注。例如维生素B12的生物合成研究,2018年张大伟课题组[218]成功实现了维生素B12在大肠杆菌中的从头合成,通过代谢调控与发酵优化使维生素B12的产量提高了250倍。2023年该课题组[219]又取得突破性进展,通过对维生素B12合成途径中30多步催化反应的整合优化,构建了维生素B12的体外无细胞催化合成系统,并实现了产量的较大提升。另外一例FDA批准的分子紫杉醇不仅在化学全合成方面取得了突破[220],在生物合成方面,2016年Ajikumar课题组[221]通过优化P450酶的表达实现了紫杉醇前体氧化型紫杉烷(570 mg/L±45 mg/L)在大肠杆菌中的高效生产。2019年王勇课题组[222]首次实现了紫杉醇关键中间体5α-羟基紫杉二烯在植物底盘中的异源合成,产量达到了1.3 μg/g水平,为实现紫杉醇未知合成途径的解析和构建紫杉醇的高效人工生物合成系统提供了可能。2021年闫建斌课题组[223]报道了红豆杉的高质量基因组数据,揭示了紫杉醇生物合成的遗传基础,为紫杉醇的合成生物学生产提供了更多可能。
在非FDA批准的药物中也有一些明星分子的生物合成研究近些年也取得了进展,如石松类生物碱的代表性生物碱石杉碱甲(huperzine A),肖友利课题组[224]发现了石杉碱甲早期生物合成步骤中催化形成中间体5-氨基戊醛的两个关键酶赖氨酸脱羧酶LDC和铜胺氧化酶CAO,随后史社坡课题组[225]又发现了生物合成石杉碱甲关键中间体石榴碱(pelletierine)的新颖聚酮合酶,并阐明了其催化机制。2023年Sattely课题组[226]发现了石松类生物碱合成过程中参与的一类新功能生物合成酶α-碳酸酐酶(CAHs),其催化立体特异性的Mannich样缩合反应以及双环骨架的形成,同时证明一系列骨架修饰步骤优化了石杉碱甲的乙酰胆碱酯酶抑制活性。另外一个明星分子银杏内酯的生物合成也取得了重要进展,2022年Pateraki课题组[227]通过对银杏内酯生物合成基因簇的挖掘和基因共表达研究,证实了左旋海松二烯是银杏内酯的关键前体,其被多个非典型的P450酶催化生成不同的银杏二萜内酯类天然产物。此外,在欧盟和澳大利亚批准上市的药物普利提环肽(plitidepsin,也称为dehydrodidemnin B)也受到了广泛关注,普利提环肽具有抗肿瘤、抗病毒和免疫抑制活性[228]。2021年,研究又发现普利提环肽可以有效抑制新冠病毒(SARS-CoV-2)的复制,抗病毒活性是实验对照药物瑞德西韦(remdesivir)的27.5倍[229],有望开发为重要的抗病毒药物。
10 展 望
通过对近十年天然产物药物的总结表明,天然产物仍然是新药发现的巨大资源宝库,涉及的天然产物结构类型也多种多样,包括类固醇、聚酮类、非核糖体肽类、萜类、生物碱类等,说明天然产物在作为治疗人类疾病的先导药物方面发挥着重要作用。尤其是近30年来,生物信息学的发展大大加快了新天然产物药物的发现速度,特别是利用丰富的基因组数据资源,快速定位到目标基因簇的核心基因,确定基因簇的边界及生物合成途径,从而将已知的活性天然产物与生物合成基因簇紧密联系起来,为进一步生物合成结构多样化的天然产物提供了帮助,例如奥利司他的半合成前体利普斯他汀、瑞扎芬净的半合成前体棘白菌素以及来法莫林的半合成前体截短侧耳素的生物合成进展,均是利用了基因组挖掘的优势而取得。同时,通过基因组挖掘可以获得很多从前难以发现的结构新颖的天然产物,例如从异小杆属线虫中发现的tapiranof和从被囊动物红树海蛸中发现的ET-743,均来自于共生生物体。在植物天然产物如依托泊苷的半合成前体鬼臼毒素、长春花碱以及秋水仙碱的生物合成研究中,转录组学和代谢组学的联合使用,同时结合组合生物合成以及异源表达手段,解决了困扰人们数十年的活性天然产物生物合成途径的解析。此外,人体及动物在特殊生理阶段所产生的内源性物质,也成为活性天然药物发现的一大来源。本综述总结了近十年天然产物药物及天然产物半合成药物前体的生物合成进展,在药物研发方面也具有很大的参考价值。首先,有助于在庞大的基因组资源中挖掘更高活性潜力化合物,通过发现并识别具有特异性结构域的核心基因或后修饰基因,进而表征得到新的骨架结构或高活性分子,或者通过保守基因的同源性挖掘,拓展分子的结构特征。其次,对于组合生物合成获得新药物类似物方面也提供了更多参考,通过对同一基因簇不同催化步骤的组合或者不同来源的同源基因簇的交叉组合,可以获得具有更加多样化的分子结构,用以探索药物的构效关系等。最后,对于建立药物的细胞生产或酶法生产路线也具有借鉴意义,包括工程化细胞中药物生成途径的搭建、前体供应途径的建立以及关键生物合成酶在药物合成中的应用等。
未来天然产物的发现仍以新颖结构和良好活性为方向,从大自然(特别是海洋和特殊环境微生物等)的“小分子宝库”中寻找新型药物或活性模板,结合全合成、组合合成和生物合成途径的操纵,将有助于促进天然产物的进一步发现,并筛选出潜在的新型候选药物。尤其近些年抗生素耐药性的蔓延,医用抗生素的疗效正在逐步下降,使得根本性的新抗生素发现迫在眉睫。现代的抗生素发现策略已经由“培养筛选-活性分析-分离鉴定”的传统方式转变为多学科、多组学(如代谢组学、蛋白质组学、转录组学、基因组学等)的联合应用策略,例如利用微生物在特殊环境里的适应性进化发现具有特殊生理意义及合成代谢途径的新抗生素(如darobactin),通过现代化的微生物培养新技术发现新的抗生素(如teixobactin),结合分子生物学和合成生物学手段挖掘基因组及激活沉默基因簇而获得新的抗生素(如macolacin)以及采取化学优化策略产生活性优异的抗生素(如G0775)等(图23)[230-233]。这些现代天然产物研究手段的组合应用,大大加快了新颖药物发现的速度。同时,新技术的出现也进一步加快了新药的发现和创新,例如在批量挖掘萜类沉默基因簇的过程中,将自动化高通量操作平台引入天然产物挖掘领域[234];CRISPR-Cas9技术在激活链霉菌沉默基因簇中的应用[235];以及在对药物分子的生物活性预测和从头设计过程中借助人工智能等[236],为处理复杂的天然产物和利用其结构获得新的创新药物开辟了新的路径。我们相信,在天然产物药物生物合成研究中的新发现,必将在针对各种人类疾病的有效药物的发现中获得实践和应用,并进一步拓展天然产物研究领域的边界。
图23
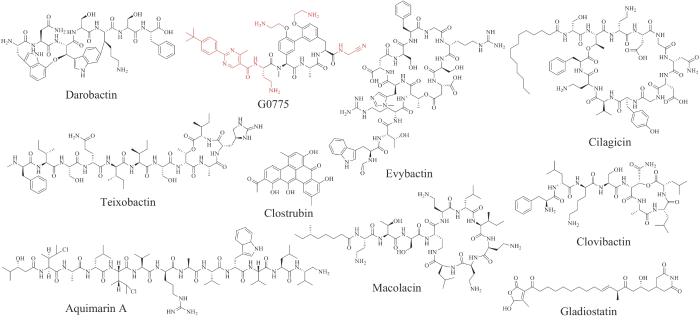
图23 近十年新发现的涉及生物合成且有发展为抗生素药物潜力的代表性天然产物

















 2897
2897

 被折叠的 条评论
为什么被折叠?
被折叠的 条评论
为什么被折叠?
 到【灌水乐园】发言
到【灌水乐园】发言









