










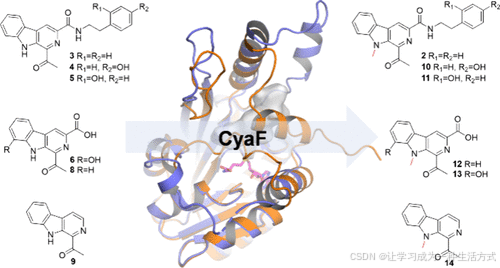
Biochemical and Structural Insights of the N-Methyltransferase CyaF in Cyanogramide Biosynthesis
N-甲基转移酶 CyaF 在 Cyanogramide 生物合成中的生化与结构见解

摘要
在天然产物生物合成中,涉及吲哚甲基化的N-甲基转移酶鲜有被发现。本研究聚焦于CyaF酶,该酶在cyanogramide生物合成过程中催化β-咔啉骨架上吲哚的关键N-甲基化步骤。从重组菌株Streptomyces coelicolor YF11/cyaABC中分离出了七种β-咔啉类似物(3–9),其中包括三种新化合物(5–7)。体外实验显示,CyaF 具有较强的底物适应性。通过解析CyaF/S-腺苷-L-高半胱氨酸(SAH)复合物的晶体结构,结合AlphaFold 预测模型及定点突变实验,揭示了CyaF 的催化机制和结构特征,阐明了其对多种底物的适应能力,并突出了其在生物催化领域的应用潜力。
生物甲基化是一种广泛存在于所有生物体中的反应,通常由S-腺苷-L-甲硫氨酸(SAM)依赖性甲基转移酶(MTs)*催化(1,2)。根据底物中*甲基受体原子的不同,MTs 可分为 O-(氧)、N-(氮)、C-(碳)和 S-(硫)取向的类型(1)。尽管 N-甲基转移酶(N-MTs) 在自然界中较为常见,但它们的数量仍少于 O-MTs(2)。然而,在天然产物生物合成中,催化吲哚甲基化的 N-MTs 仍然极为罕见(图 1A)。首个吲哚 N-MT 于 1982 年 在兔肝中被发现(3)。在植物中,已有三种 γ-生育酚 N-MTs 被报道,它们分别参与ajmaline(阿马林)、desacetoxyvindoline(去乙酰氧长春胺)*和 **ervincine** 的吲哚基团 **N-甲基化**(4,5)。其中,**阿马林(Ajmaline)** 是一种用于治疗*室性和房室性心律失常的临床抗心律失常药物,其生物合成的最后一步便是 N-甲基化(6)。去乙酰氧长春胺(Desacetoxyvindoline) 是长春碱(vindoline)*生物合成中的重要中间体,而长春碱是*抗癌药长春新碱(vinblastine)和长春花碱(vincristine)的前体(7)。在蓝细菌中,WelM 被证明可催化 氧吲哚(oxindole)*的 **N-甲基化**,参与 **N-甲基-welwitindolinone C 异硫氰酸酯**的生物合成(8)。该化合物已被证实可*降低 MCF-7/ADR 细胞对基于天然产物的抗癌药物(包括长春新碱、紫杉醇、放线菌素 D、柔红霉素和秋水仙碱等)的耐药性(9,10)。迄今为止,在放线菌中,仅有CyaF 被鉴定为参与cyanogramide生物合成的吲哚 N-甲基转移酶(1)(图 1B)。
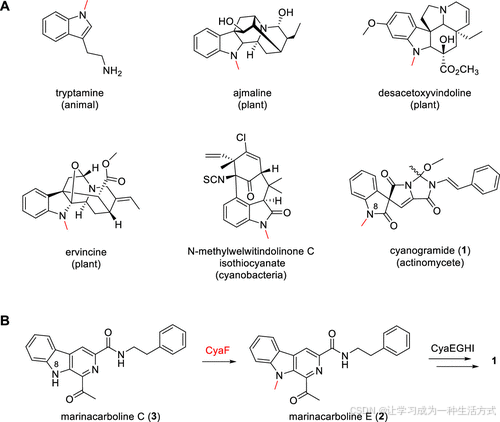
图1. 代表性的吲哚 N-甲基化天然产物(A)和N-甲基转移酶 CyaF在cyanogramide (1)生物合成中作用的推测功能(B)。
Cyanogramide (1) 首次从海洋来源的 Actinoalloteichus cyanogriseus WH1-2216-6 中分离出,特征是一个吡咯[1,2-c]咪唑并[4,5-d]吡咯结构,该结构与氧吲哚螺环融合(11)。化合物 1 在K562/A02 和 MCF-7/Adr 细胞中有效逆转了阿霉素耐药性,并在 KB/VCR 细胞中逆转了长春新碱的耐药性(11)。它还展示了对人类胶质瘤 U251 和 U87MG 细胞的细胞毒性活性(12)。此外,化合物 1 显示出治疗炎症性肠病的潜力(13,14)。最近,我们通过异源表达在Streptomyces coelicolor YF11中表征了A. cyanogriseus WH1-2216-6中cyanogramide (1) 的生物合成基因簇(cya),并基于体内基因失活和喂养实验提出了简明的生物合成途径(图 S1)(15)。在前期研究中,MT CyaF 被认为催化了β-咔啉骨架中吲哚部分的 N-甲基化,将marinacarboline C (3) 转化为 marinacarbolines E (2)(图 1B)(15)。然而,CyaF 的生化活性仍未得到证明。
在本研究中,从重组菌株 S. coelicolor YF11/cyaABC 中分离出七种β-咔啉类似物(3–9),包括三种新化合物(5–7)。通过体外酶学分析,对N-MT CyaF的生化特性进行了表征,显示其具有底物适应性。此外,结合结构分析和定点突变实验,揭示了CyaF的催化机制,并为其接受替代底物的能力提供了结构基础,突出了其在生物催化应用中的潜力。
结果与讨论
CyaF 底物的制备
生物信息学分析和体内基因破坏表明,cyaF 基因应编码一种 N-MT,能够通过将甲基团引入到N-8位,催化marinacarboline C (3) 转化为 marinacarboline E (2)(图 1B)(15)。在先前提出的 cyanogramide (1) 生物合成途径中(图 S1),marinacarboline C (3) 骨架由三种酶合成:脂肪酸 CoA 连接酶(CyaA)、Pictet–Spengler 环化酶(CyaB) 和 谷氨酸脱羧酶(CyaC)(15)。为了获得更多的底物用于CyaF的功能研究,将三个基因cyaA、cyaB 和 cyaC 一起引入 S. coelicolor YF11 中,得到重组菌株 S. coelicolor YF11/cyaABC。该菌株产生了三种新化合物(5–7),以及四种已知化合物,3、marinacarboline B (4)、1-乙酰-3-羧基-β-咔啉 (8) 和 1-乙酰-β-咔啉 (9)(图2)。通过比较它们的 HRESIMS 和 NMR 数据与已报道的数据,对已知化合物进行了结构鉴定(图 S2–S9)(16,17)。通过对新化合物 5–7 进行广泛的 HRESIMS 和 1D、2D NMR 数据分析(图 S10–S36 和表 S1),确定了它们的结构。提出的 3–9 化合物的生物合成途径如图 S37 所示。
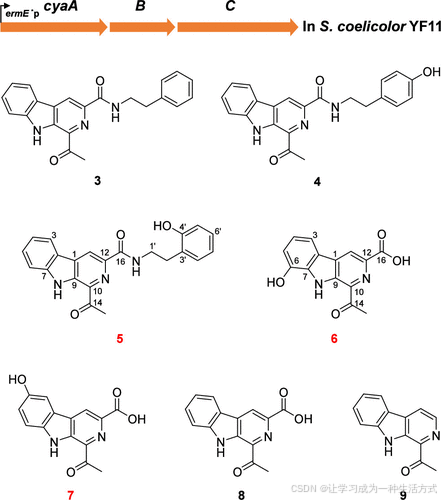
图2. 从重组菌株 S. coelicolor YF11/cyaABC 中分离出的化合物 3–9 的结构。
Marinacarboline R (5) 以白色无定形粉末形式分离得到。HRMS 分析确定其分子式为 C22H19N3O3 (m/z 396.1322 [M + Na]+,计算值为 C22H19N3O3Na 396.1318),表明与 marinacarboline C (3) 相比,5 中含有一个额外的氧原子 (m/z 358.1551 [M + H]+,计算值为 C22H20N3O2 358.1550)。对 5 的 NMR 数据分析揭示其与化合物 3 相似(16)。主要区别在于,3 中的 H-4′ (δH = 7.34) 的 1H NMR 信号在 5 中缺失。此外,5 中 C-4′ 的 碳信号 从 3 中的 δC 129.0 去屏蔽至 δC 155.5,表明在 5 的 C-4′ 位置存在一个 羟基。通过从 H-5′/H-6′/H-8′ 到 C-4′ 的 HMBC 相关性,确认了 5 中的 4′-OH。 因此,化合物 5 被表征为 3 的 C-4′ 羟基化衍生物(图 2)。
Marinacarboline S (6) 以黄色无定形粉末形式分离得到。通过 HRMS 分析,确定其分子式为 C14H10N2O4 (m/z 293.0537 [M + Na]+,计算值为 C14H10N2O4Na 293.0532)。化合物 6 展示了与 dichotomine F 相似的 1H 和 13C NMR 信号(18),唯一的不同是缺少 dichotomine F 中甲基酯的信号。此外,C-16 的信号从 dichotomine F 中的 δC 165.2 去屏蔽到 6 中的 δC 167.0。根据这些数据,6 被阐明为 dichotomine F 的 去甲基衍生物(图 2)。
HRMS 分析表明,marinacarboline T (7) 和 6 具有相同的分子式。对 NMR 数据的详细比较显示,7 和 6 的结构高度相似。关键区别在于,7 中含有 4-OH,而 6 中含有 6-OH。7 中 4-OH 的定位得到了 H-5/H-6 之间的 COSY 相关性,以及 H-3 到 C-1/C-5/C-7、H-5 到 C-3/C-4/C-7、H-6 到 C-2/C-4 的 HMBC 相关性的支持。
CyaF 的生化特性
可溶性 N-His6 标签 CyaF 蛋白在携带质粒 pSCG2304 的 E. coli BL21(DE3) 中表达(表 S2),并通过 Ni-NTA 色谱纯化至近乎纯度(图 S38)。在含 SAM 的生化实验中,重组 CyaF 确实将 3 转化为 2,且 2 的产生随着孵育时间的延长而增加(图 3A 和图 S39)。然而,在缺乏 CyaF 或 SAM 的对照实验中没有观察到 3 转化为 2(图 3A)。这些数据证实了 CyaF 作为一种特异性的 N-MT。
随后,通过使用化合物 4–9 研究了 CyaF 的底物特异性。化合物 4、5 和 8 在 5 μM CyaF 的作用下高效地转化为各自的甲基化产物 10、11 和 12(图 3B 和图 S40–S42),而化合物 6 和 9 转化为预期产物 13 和 14(图 3C)则需要更高浓度的 CyaF(50 μM)(图 S43,S44)。即使在 100 μM CyaF 的情况下,化合物 7 也未观察到转化(图 3C 和图 S45),这表明 4-OH 可能会妨碍 7 与 CyaF 的结合。结果表明,CyaF 显示了一定程度的底物适应性。
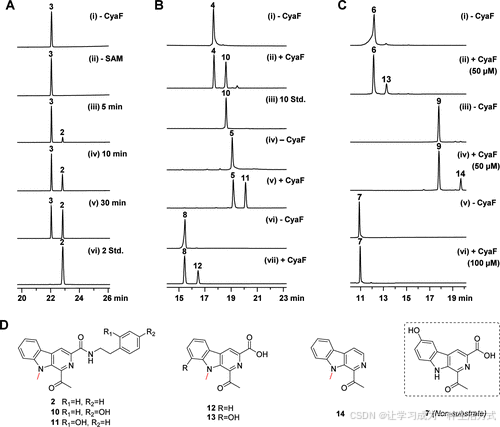
图3. CyaF催化反应的体外表征。 (A) HPLC 分析 CyaF(5 μM)与底物 3 的时间过程实验: (i) 不含 CyaF 作为对照; (ii) 不含 SAM 作为对照; (iii) 5 分钟; (iv) 10 分钟; (v) 30 分钟; (vi) 标准化合物 2。
(B) HPLC 分析 CyaF 与底物 4、5 和 8(5 μM CyaF,孵育 1 小时)。
(C) HPLC 分析 CyaF 与底物 6、9 和 7(对于 6 和 9 使用 50 μM CyaF,对于 7 使用 100 μM CyaF,孵育 1 小时)。
(D) CyaF 与底物 3–6、8 和 9 形成的产物结构。
CyaF 的结构与底物识别
为了了解 CyaF 的底物结合模式,我们首先使用 AlphaFold3 构建了一个高置信度的 CyaF 模型(图 4A)。该模型与 Anabaena variabilis ATCC 29413 中的 MT(PDB ID: 3ggd,CyaF 与其序列相似度为 28%,由 Joint Center for Structural Genomics 解析)相似(图 S46)。值得注意的是,包括 α7 螺旋、23–40 区段的环 和 β6–7 链之间的环(氨基酸残基 219–230)的区域(图 4A)有助于形成一个底物结合口袋,该口袋与底物 3 的形状紧密匹配。因此,我们成功地将底物 3–5 对接到 CyaF 模型的活性口袋中,其中β-咔啉平面紧密地适应于平坦的口袋,而苯乙胺和酪胺部分则朝向口袋入口(图 4B)。这种结合模式将 β-咔啉 的 N8 原子与 SAM 的甲基团的距离放置在约 3.4 Å(图 4C),这一距离与已报道的 SAM 甲基化反应中的距离一致。(2)
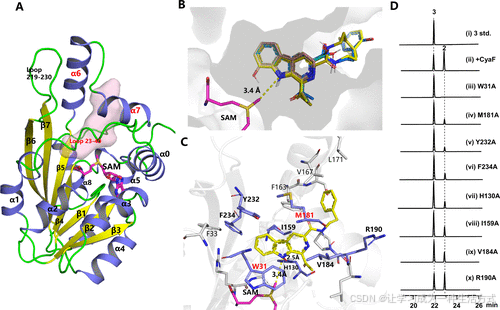
图 4. CyaF 的底物识别 (A) 由 AlphaFold3 预测的 CyaF 结构模型。CyaF 模型中的底物口袋由 POCASA 1.1 (19) 预测,并以粉色表面显示。 (B) 底物 3–6、8 和 9 能够有效地对接并在 CyaF 结构模型的活性口袋内排列。 (C) 3 号化合物在 AlphaFold3 预测的 CyaF 结构模型中的结合模式。 (D) CyaF 的突变结果。
有趣的是,在对接模型中,位于 23–40 号环上的 W31 残基以及 α7 螺旋上的 M181 残基分别与 β-咔啉平面和酰基基团相互作用(图 4C)。与对接模型一致,W31A 突变体表现出几乎完全丧失的活性,而 M181A 突变体的活性显著降低(图 4D)。对 CyaF 与 3 号化合物的对接模式进行更详细的分析表明,Y232、F234 和 H130 在结合过程中起着关键作用。β-咔啉核心的 N8 原子与 H130 侧链形成氢键,而 Y232A 和 F234A 通过 T−π 相互作用稳定 β-咔啉平面。Y232A、F234A 和 H130A 突变进一步验证了预测的 CyaF 结构及其结合模式(图 4D)。此外,用丙氨酸替换 β-咔啉基团附近的 I159 和 V184 会适度降低活性,表明它们参与了疏水相互作用。相比之下,作为阴性对照的 R190A 突变体保留了几乎与野生型酶相同的活性,支持 R190 并不参与底物结合的结论(图 4D)。
基于该 CyaF 模型,我们还研究了化合物 6–9 的结合情况。化合物 6、8 和 9 的 β-咔啉核心在 CyaF 活性口袋中的定位方式与化合物 3–5 类似(图 S47)。然而,6、8 和 9 号化合物缺乏苯乙胺和酰基基团,无法与 F163、V167、L171 和 M181 残基相互作用,这可能导致 CyaF 对这些化合物的催化效率降低。值得注意的是,化合物 7 采用不同的取向,其 C4-OH 基团可能会受到活性位点残基的空间位阻,从而解释了其活性缺失的现象。
可能的底物诱导 CyaF 构象重排
为了进一步阐明酶促反应机制和底物的适应性,我们尝试将底物 3 共结晶或浸泡于 CyaF 的活性位点中。然而,我们仅解析出了 CyaF 与 S-腺苷-L-高半胱氨酸(SAH)复合物的晶体结构,分辨率为 2.7 Å(PDB ID: 9K2F,表 S3)。不对称单元中包含两个 CyaF 单体,每个单体均采用 Rossmann 折叠(α–β–α 夹层),这是 I 类甲基转移酶(MTs)的特征结构 (2)(图 5A 和 5B)。在这两个单体中,SAH 分子均结合于 Rossmann 折叠结构中,并具有清晰的电子密度(图 5A)。
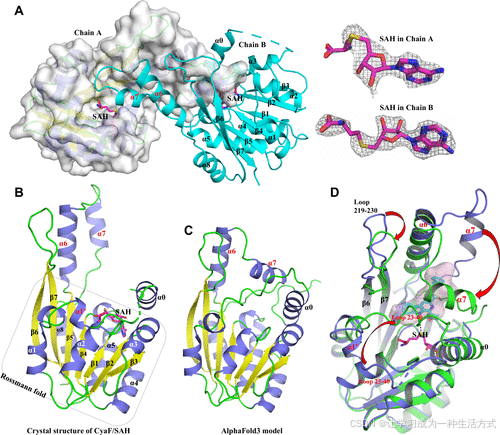
图 5. CyaF 与 SAH 复合物的晶体结构。
(A) 在不对称单元中存在两个 CyaF 单体,其中链 B 的 α6–7 螺旋嵌入链 A 的活性腔,与链 A 的 β6–7 链密切相互作用。在两个链中,SAH 分子均结合在 Rossmann 折叠中,其 2FO–FC 电子密度图(1.5σ 等值)以灰色网格显示。 (B) CyaF 单体的示意图表示。 (C) 由 AlphaFold3 预测的 CyaF 结构模型。 (D) 解析出的 CyaF/SAH 结构(蛋白质以紫色表示,SAH 以紫罗兰色表示)与 AlphaFold3 预测模型(绿色)的叠加,揭示了 CyaF 的 α7 螺旋、23–40 段环和 β6–7 链之间的环(残基 219–230)发生构象变化。
有趣的是,将实验解析的 CyaF/SAH 结构与 AlphaFold 预测模型(图 5C)进行比较时,发现 Rossmann 折叠域高度保守,但 CyaF 的 α7 螺旋、23–40 段环以及 β6–7 链之间的环(残基 219–230)在构象上存在显著差异(图 5D)。在 CyaF/SAH 结构中,这些区域远离 SAH,而在 AlphaFold 预测模型中,这些区域朝向 SAM,形成与底物形状高度匹配的底物结合口袋。由于多种甲基转移酶在结合底物时会经历结合口袋的构象变化(参考文献 20–22),我们推测 CyaF 的 α7 螺旋、23–40 段环和 219–230 残基环可能发生显著的构象重排,以转变为结合底物后的“闭合”活性状态。而我们解析的 CyaF/SAH 结构可能代表“开放”状态。在“开放”构象中,不对称单元内的一个 CyaF 单体的 α6–7 螺旋伸入另一个单体的活性腔,完全占据了 SAH 附近的拟议催化中心(图 5A)。这种堆积排列可能有助于 CyaF 蛋白的结晶,但也阻碍了我们获得 CyaF–底物复合物的结构。
结论
总之,我们分离了三种新的 β-咔啉生物碱,并证明 CyaF 是一种 N-甲基转移酶(N-MT),通过 SAM 作为甲基供体催化氰蓝酰胺生物合成中的 N8 甲基化。CyaF 在体外实验中可作用于多种底物,这些底物均具有相似的 β-咔啉骨架,但侧链不同,并产生了多种甲基修饰的衍生物。此外,分子对接和定点突变实验支持 CyaF 存在“闭合构象状态”,并解释了其对底物的宽泛选择性。我们解析了 CyaF 与 SAH 复合物的晶体结构,揭示了相较于 AlphaFold 预测模型的“开放构象状态”。本研究为 β-咔啉吲哚部分的甲基化提供了重要见解,同时也展示了 AlphaFold 在克服实验技术局限性方面的潜力,表明基于 AI 的预测可以提供有关酶机制的更动态的视角。
实验部分
一般实验方法
NMR 光谱在 Avance 500 谱仪(Bruker)上测得,¹H 核的测量频率为 500 MHz,¹³C 核的测量频率为 125 MHz。化学位移 (δ) 以 TMS 信号为参考。高分辨质谱数据采用 Bruker Maxis 4G UHR-TOFMS 质谱仪(Bruker Daltonics Inc.)测定。柱层析 (CC) 使用硅胶 (100–200 目,江油硅胶发展有限公司) 进行。半制备高效液相色谱 (HPLC) 采用 Primaide 系统(日立公司)和 ODS-A 柱 (10 × 250 mm, 5 μm, YMC) 进行分离。所有 PCR 验证均使用 DL2000 Plus DNA marker(Takara Bio Inc.)和 EasyTaq DNA 聚合酶(全式金生物技术有限公司)。所有化学试剂和溶剂均为分析纯或色谱纯。
细菌菌株与质粒
本研究中使用或构建的所有菌株和质粒均列于表 S2。重组菌株 S. coelicolor YF11/pCSG2315 (cyaABC) 在 28°C 下分别于孢子培养基(20 g 大豆粉、20 g 甘露醇、20 g 琼脂粉、1 L 蒸馏水,pH 7.0)和发酵培养基(20 g 葡萄糖、3 g 牛肉膏、10 g 酵母膏、10 g 淀粉、10 g 可溶性蛋白胨、0.5 g K₂HPO₄、0.5 g MgSO₄、2 g CaCO₃、1 L 蒸馏水,pH 7.0)中培养。大肠杆菌菌株在 37°C 下于液体 LB 培养基中培养。
发酵、提取及代谢产物分析
为分离化合物 3–9,共培养 S. coelicolor YF11/pCSG2315 (cyaABC) 15 L,培养温度 28°C,摇床转速 200 rpm,培养 6 天。发酵液经离心(3900 rpm, 12 min)分离出上清和菌丝体。上清用等体积丁酮提取三次,菌丝体用 2 L 丙酮提取三次,减压浓缩后,残余物重新用 2 L 丁酮提取四次,最终获得 1.77 g 提取物。提取物经 正相硅胶柱层析 (NPSC)(100–200 目)分离,以 CHCl₃/MeOH 梯度洗脱(1:0、4:1、2:1、0:1,v/v,80 mL)得到四个部分 (Fr1–Fr4)。
-
Fr1 经 NPSC(200–300 目) 再分离,环己烷/乙酸乙酯梯度洗脱(1:0、1:1、0:1,v/v,120 mL),得 5 个亚组分 (Fr1.1–Fr1.5)。其中 Fr1.2 和 Fr1.3 通过半制备 HPLC 分离,分别得到 化合物 9(4.0 mg) 和 化合物 5(5.4 mg)。
-
Fr2 采用 Sephadex LH-20 层析,以 CHCl₃/MeOH (1:1, v/v) 洗脱,得到 4 个亚组分 (Fr2.1–Fr2.4)。Fr2.1 和 Fr2.4 通过半制备 HPLC 纯化,分别获得 化合物 3(3.5 mg) 和 化合物 4(2.5 mg)。
-
Fr3 进一步采用 Sephadex LH-20 分离,CHCl₃/MeOH (1:1, v/v) 洗脱,得到 4 个亚组分 (Fr3.1–Fr3.4)。Fr3.3 和 Fr3.4 通过半制备 HPLC 纯化,分别获得 化合物 6(1.7 mg) 和 化合物 7(1.8 mg)。对 Fr3.4 进一步纯化,获得 化合物 8(3.7 mg)。
化合物数据
-
化合物 5(白色粉末):UV (MeOH) λmax 375 nm (4.08), 286 nm (5.09), 204 nm (5.19); IR (film) νmax 3342.64, 2922.16, 2839.33, 1635.64, 1593.20, 1454.33, 1020.34, 754.17; HRESIMS: m/z 396.1322 [M + Na]⁺ (C₂₂H₁₉N₃O₃Na, 396.1318)。
-
化合物 6(黄色粉末):UV (MeOH) λmax 278 nm (4.05), 228 nm (5.17); IR (film) νmax 3415.93, 1653.00, 1436.97, 1406.11, 1315.45, 1014.56, 950.91, 704.02; HRESIMS: m/z 293.0537 [M + Na]⁺ (C₁₄H₁₀N₂O₄Na, 293.0532)。
-
化合物 7(绿色粉末):UV (MeOH) λmax 300 nm (4.27), 229 nm (5.19); IR (film) νmax 3437.15, 1660.71, 1433.11, 1409.96, 1018.41, 950.91, 700.16; HRESIMS: m/z 293.0533 [M + Na]⁺ (C₁₄H₁₀N₂O₄Na, 293.0532)。
CyaF 的克隆、过表达及纯化
基因 cyaF 由 A. cyanogriseus WH1-2216-6 基因组 DNA 通过 PCR 扩增,使用 NdeI/BamHI 酶切并连接至 pET28a 载体,构建 pCSG2304。pCSG2304 转入 E. coli BL21(DE3) 表达 His₆-CyaF 融合蛋白。 诱导表达后,菌体收集并超声裂解,Ni-NTA 亲和层析纯化,最终在 −80°C 存储。CyaF 产量约 50 mg/L。
CyaF 的定点突变及纯化
构建 W31A, H130A, I159A, M181A, V184A, R190A, Y232A, F234A 突变体,采用与 WT CyaF 相同方法纯化,产量约 40–50 mg/L。
CyaF 及突变体的生化分析
在 PBS 缓冲液 (50 mM, pH 7.0) 中,CyaF 与底物 3–9 反应,条件为 30°C,含 200 μM 底物、5 μM(或 50、100 μM)CyaF、1 mM SAM、1% DMSO、5% 甘油。
分子对接
利用 AutoDock Vina 对接底物 3–9,几何约束由 Grade Web Server 生成。
数据可用性:最终结构坐标及结构因子已存入 PDB,图像由 PyMOL 绘制。

















 2951
2951

 被折叠的 条评论
为什么被折叠?
被折叠的 条评论
为什么被折叠?
 到【灌水乐园】发言
到【灌水乐园】发言









