






目录
1. 加载数据集,创建Seurat对象
数据下载地址:https://cf.10xgenomics.com/samples/cell/pbmc3k/pbmc3k_filtered_gene_bc_matrices.tar.gz
Pbmc:外周血单核细胞,包括淋巴细胞(T细胞,B细胞和自然杀伤(NK)细胞)和单核细胞,一小部分的树突状细胞。
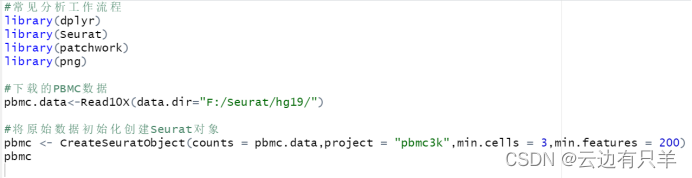
其中CreateSeuratObject()函数用来创建Seurat对象
counts:一个类似矩阵的对象;
project:Seurat对象的项目名称;
min.cells:包含至少在这么多细胞中检测到的特征;
min.features:包含至少可以检测到这些特征的单元格。
运行结果:

2. 数据预处理
a. QC(质量控制)
三个标准
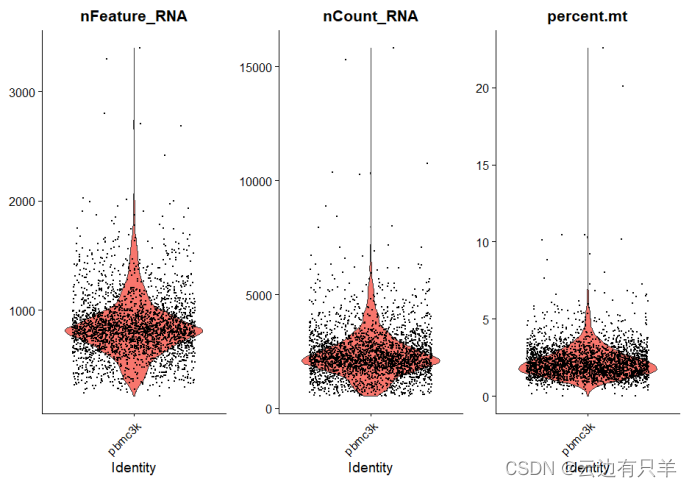
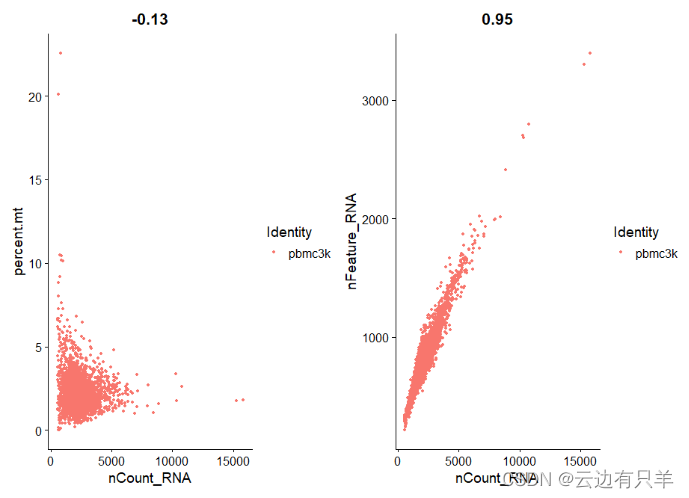
1. 每个细胞中检测到的唯一基因数(nFeature_RNA)
- 低质量的细胞或空的droplet液滴通常含有很少的基因
- Cell doubles 或 multiplets可能表现出异常高的基因计数
2. 每个细胞中检测到的总分子数(nCount_RNA)
3. 线粒体基因含量比例(percent.mt)
- 低质量或者死亡细胞含有很高的线粒体基因
- 使用PercentageFeatureSet()计算线粒体QC比例
- MT-开头的基因是线粒体基因
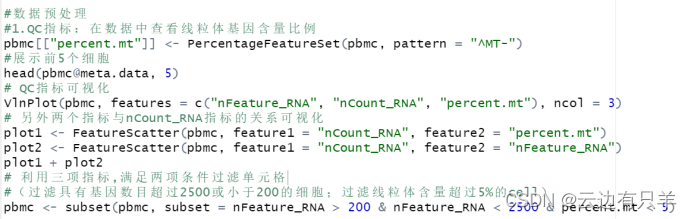
运行结果:

b. 数据标准化
默认标准化方法为LogNormalize,标准化后的数据存在pbmc[[“RNA”]]@data中
归一化 -> 乘以比例因子10000 -> 对数转换
![]()
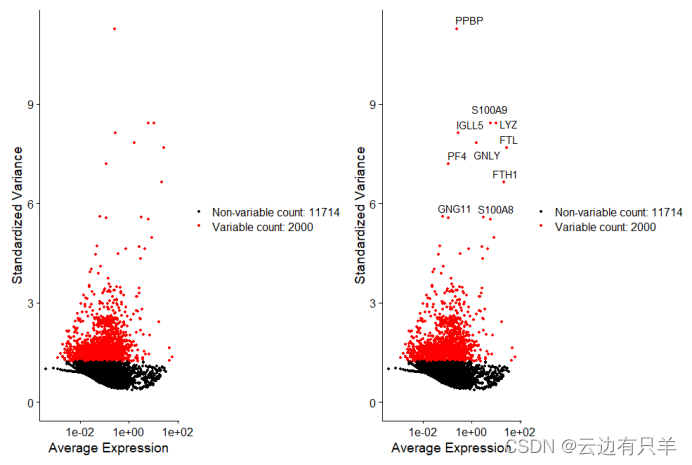
c. 高变基因(特征选择)
选择数据集中高变异的特征子集(在某些cell中高表达,在某些cell中低表达)
通过FindVariableFeatures()函数,计算每一个基因的均值和方差,默认选择高变异的2000个基因用于下游分析

运行结果:
![]()
d. 归一化
使用ScaleData()函数转化(“归一化”)数据,使每一个基因在所有cell中的表达均值为0,方差为1
结果在pbmc[[“RNA”]]@scale.data中,通过str()获取pbmc的数据结构

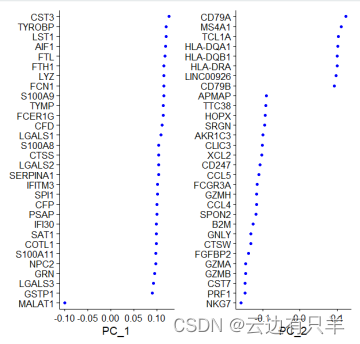
e. 数据降维(PCA)

运行结果:

Positive和Negative就是PC轴的正负映射关系,正值为Positive,负值为Negative。返回的是正值和负值绝对值最大的top30。可以理解为对所有细胞区分度最大的基因。
运行完RunPCA后,得到两个分解矩阵,2000x50的权重矩阵,50x2000的系数矩阵,总共为50个PC

结果可视化

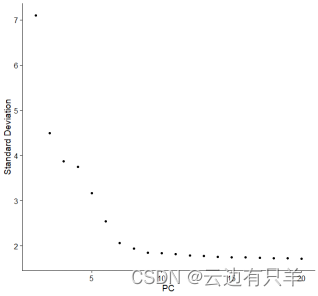
确定维度
ElbowPlot(pbmc)是确定用多少个PC进行分群的一种有效方法,直观地显示了每个PC的标准偏差,我们要找的就是标准偏差趋于平稳的位置
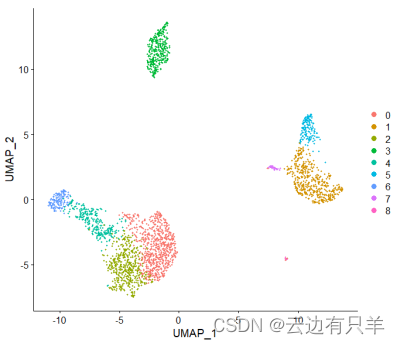
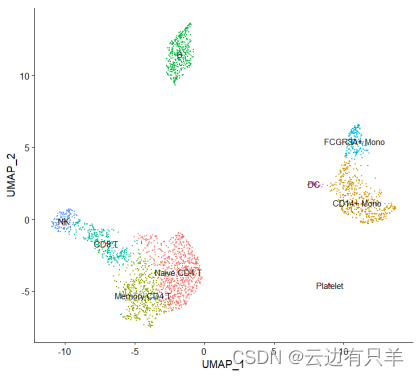
3. 单细胞聚类
首先根据PCA空间中的欧氏距离构建KNN图,并根据其局部邻域中的共享重叠(jaccard相似性)细化任意两个单元之间的边权重。共分为9类

前5个细胞所属的类别为:

4. 非线性降维
首先下载UMAP算法,进行可视化

运行结果:
5. 细胞类型标识分配给集群
用新的标识替代原来的cluster0-8

运行结果:
6. 结果保存为.rds文件
saveRDS(pbmc, file = "F:/Seurat/1_1.rds")

















 1065
1065

 被折叠的 条评论
为什么被折叠?
被折叠的 条评论
为什么被折叠?
 到【灌水乐园】发言
到【灌水乐园】发言









