







「写在前面」
学习一个软件最好的方法就是啃它的官方文档。本着自己学习、分享他人的态度,分享官方文档的中文教程。软件可能随时更新,建议配合官方文档一起阅读。推荐先按顺序阅读往期内容:
文献篇:
1.文献阅读:(Seurat V1) 单细胞基因表达数据的空间重建
2.文献阅读:(Seurat V2) 整合跨越不同条件、技术、物种的单细胞转录组数据
3.文献阅读:(Seurat V3) 单细胞数据综合整合
4.文献阅读:(Seurat V4) 整合分析多模态单细胞数据
5.文献阅读:(Seurat V5) 用于集成、多模态和可扩展单细胞分析的字典学习
教程篇:
1.Seurat Tutorial 1:常见分析工作流程,基于 PBMC 3K 数据集
2.Seurat Tutorial 2:使用 Seurat 分析多模态数据
3.Seurat Tutorial 3:scRNA-seq 整合分析介绍
4.Seurat Tutorial 4:映射和注释查询数据集
5.Seurat Tutorial 5:使用 reciprocal PCA (RPCA) 快速整合
6.Seurat Tutorial 6:整合大型数据集的技巧
7.Seurat Tutorial 7:整合 scRNA-seq 和 scATAC-seq 数据
目录
-
1 介绍:Seurat v4 Reference Mapping -
2 示例1:映射人类外周血细胞图谱 -
2.1 一个多模态 PBMC 参考数据集 -
2.2 映射 -
2.3 探索映射结果 -
2.4 计算新的 UMAP 可视化
-
-
3 示例2:映射人类骨髓细胞图谱 -
3.1 多模态 BMNC 参考数据集 -
3.2 计算 sPCA 变换 -
3.3 计算缓存的邻居索引 -
3.4 查询数据集预处理 -
3.5 映射 -
3.6 探索映射结果
-
官网教程:
https://satijalab.org/seurat/articles/multimodal_reference_mapping
1 介绍:Seurat v4 Reference Mapping
此小节介绍了在 Seurat 中将 query 数据集映射到注释的 references 中的过程。在此示例中,我们将 10X Genomics 发布的 2,700 个 PBMC 的首批 scRNA-seq 数据集之一映射到我们最近描述的使用 228 种抗体测量的 162,000 个 PBMC 的 CITE-seq reference。我们选择这个例子是为了演示 reference 数据集指导下的监督分析如何帮助枚举细胞状态,而使用无监督分析很难找到这些细胞状态。在第二个示例中,我们演示了如何将来自不同个体的人类 BMNC 的人类细胞图谱数据集连续映射到一致的 reference 上。
我们之前演示了如何使用 reference-mapping 方法来注释 query 数据集中的细胞标签。在 Seurat v4 中,我们大幅提高了集成任务(包括 reference mapping)的速度和内存要求,并且还包含将 query cells 投影到先前计算的 UMAP 可视化上的新功能。
在此小节中,我们演示了如何使用先前建立的 reference 来解释 scRNA-seq query:
-
根据一组 reference 定义的细胞状态注释每个 query 细胞 -
将每个 query 细胞投影到先前计算的 UMAP 可视化上 -
估算 CITE-seq reference 中测量的表面蛋白的预测水平
要运行此小节,请安装 CRAN 上提供的 Seurat v4。此外,您还需要安装 SeuratDisk 软件包。
install.packages("Seurat")
remotes::install_github("mojaveazure/seurat-disk")
library(Seurat)
library(SeuratDisk)
library(ggplot2)
library(patchwork)
2 示例1:映射人类外周血细胞图谱
2.1 一个多模态 PBMC 参考数据集
我们从最近的论文中加载 reference(download here),并可视化预先计算的 UMAP。该 reference 存储为 h5Seurat 文件,这种格式支持多模态 Seurat 对象的磁盘存储(有关 h5Seurat 和 SeuratDisk 的更多详细信息可以在此处找到)。
reference <- LoadH5Seurat("../data/pbmc_multimodal.h5seurat")
DimPlot(object = reference, reduction = "wnn.umap", group.by = "celltype.l2", label = TRUE, label.size = 3, repel = TRUE) + NoLegend()
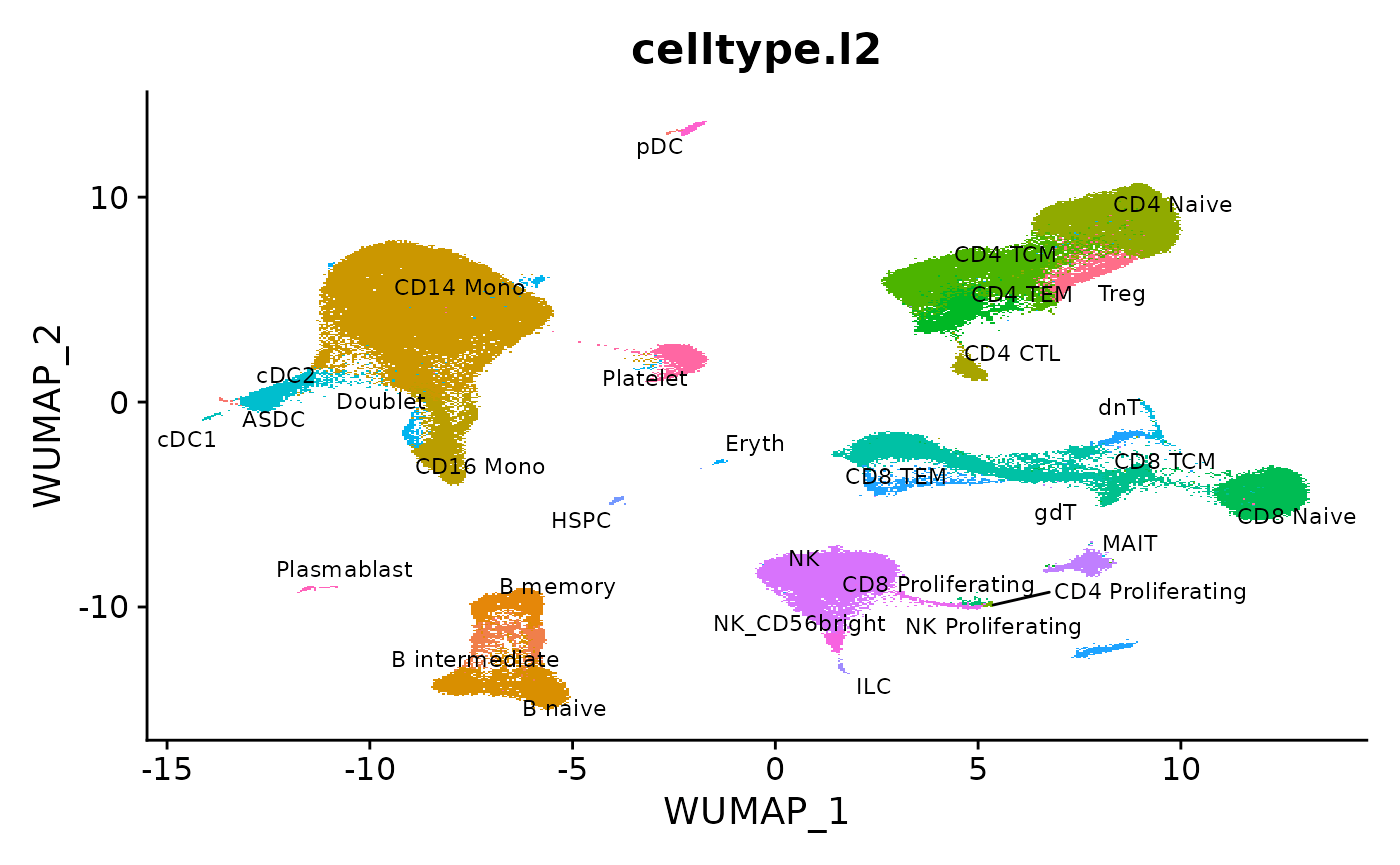
2.2 映射
为了演示与此多模态 reference 的映射,我们将使用由 10x Genomics 生成并可通过 SeuratData 获取的 2,700 个 PBMC 数据集。
library(SeuratData)
InstallData('pbmc3k')
reference 是使用 SCTransform() 进行归一化,因此我们在这里使用相同的方法对 query 进行归一化。
pbmc3k <- SCTransform(pbmc3k, verbose = FALSE)
然后我们找到 reference 和 query 之间的 anchors。如手稿中所述,我们在此示例中使用了预先计算的 supervised PCA (spca) 转换。我们建议对 CITE-seq 数据集使用 supervised PCA,并在此节的下一栏演示如何计算此转换。但是,您也可以使用标准 PCA 转换。
anchors <- FindTransferAnchors(
reference = reference,
query = pbmc3k,
normalization.method = "SCT",
reference.reduction = "spca",
dims = 1:50
)
然后,我们将细胞类型标签和蛋白质数据从 reference 转移到 query。此外,我们将 query 数据投影到 reference 的 UMAP 结构上。
pbmc3k <- MapQuery(
anchorset = anchors,
query = pbmc3k,
reference = reference,
refdata = list(
celltype.l1 = "celltype.l1",
celltype.l2 = "celltype.l2",
predicted_ADT = "ADT"
),
reference.reduction = "spca",
reduction.model = "wnn.umap"
)
MapQuery 在做什么?
MapQuery() 是三个函数的包装:TransferData()、IntegrateEmbeddings() 和 ProjectUMAP()。TransferData() 用于传输细胞类型标签并估算 ADT 值。IntegrateEmbeddings() 和 ProjectUMAP() 用于将 query 数据投影到 reference 的 UMAP 结构上。使用中间函数执行此操作的等效代码如下:
pbmc3k <- TransferData(
anchorset = anchors,
reference = reference,
query = pbmc3k,
refdata = list(
celltype.l1 = "celltype.l1",
celltype.l2 = "celltype.l2",
predicted_ADT = "ADT")
)
pbmc3k <- IntegrateEmbeddings(
anchorset = anchors,
reference = reference,
query = pbmc3k,
new.reduction.name = "ref.spca"
)
pbmc3k <- ProjectUMAP(
query = pbmc3k,
query.reduction = "ref.spca",
reference = reference,
reference.reduction = "spca",
reduction.model = "wnn.umap"
)
2.3 探索映射结果
我们现在可以可视化 2,700 个 query cells。它们已被投影到 reference 定义的 UMAP 可视化中,并且每个都已收到两个粒度级别的注释(level 1 和 level 2)。
p1 = DimPlot(pbmc3k, reduction = "ref.umap", group.by = "predicted.celltype.l1", label = TRUE, label.size = 3, repel = TRUE) + NoLegend()
p2 = DimPlot(pbmc3k, reduction = "ref.umap", group.by = "predicted.celltype.l2", label = TRUE, label.size = 3 ,repel = TRUE) + NoLegend()
p1 + p2
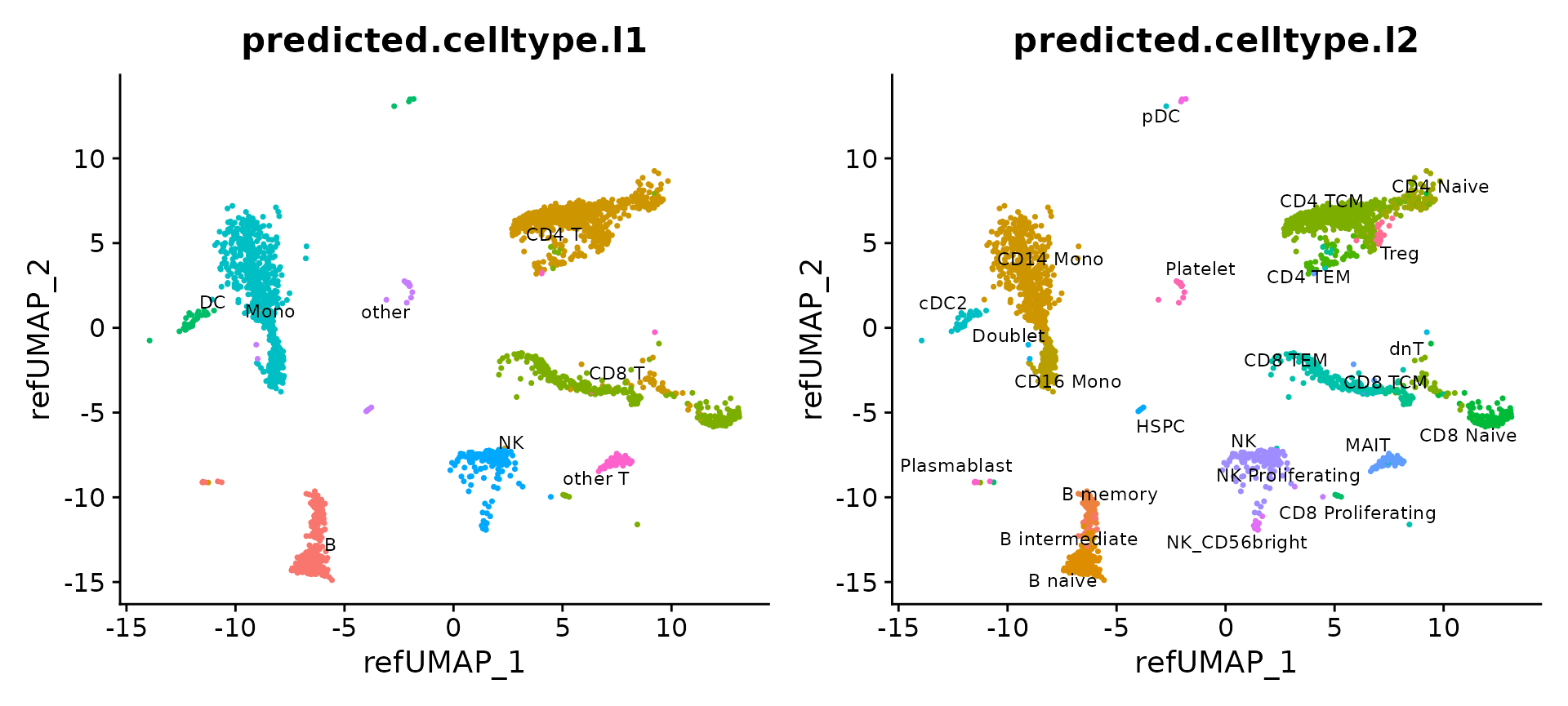
reference-mapped 数据集帮助我们识别先前在 query 数据集的无监督分析中混合的细胞类型。仅举几个例子,包括 plasmacytoid dendritic cells (pDC), hematopoietic stem and progenitor cells (HSPC), regulatory T cells (Treg), CD8 Naive T cells, cells, CD56+ NK cells, memory, and naive B cells, and plasmablasts。
每个预测都会分配一个 0 到 1 之间的分数。
FeaturePlot(pbmc3k, features = c("pDC", "CD16 Mono", "Treg"), reduction = "ref.umap", cols = c("lightgrey", "darkred"), ncol = 3) & theme(plot.title = element_text(size = 10))
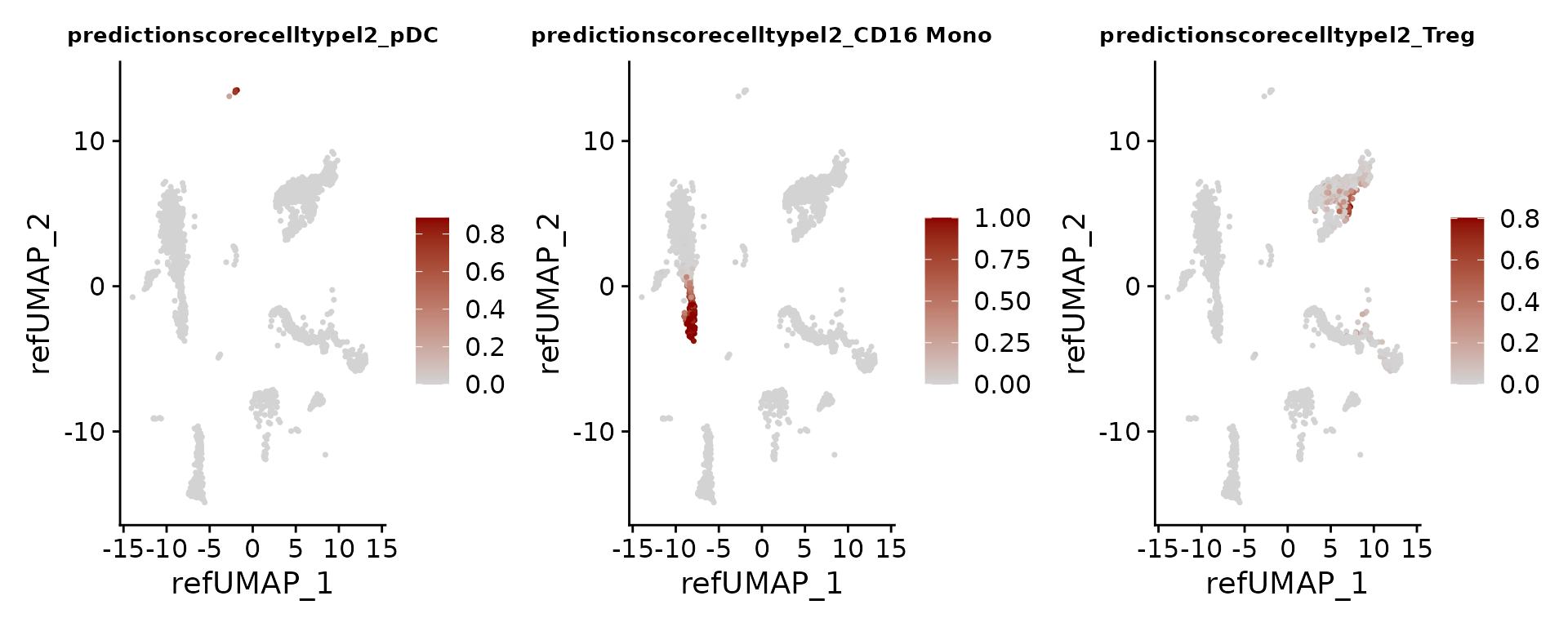
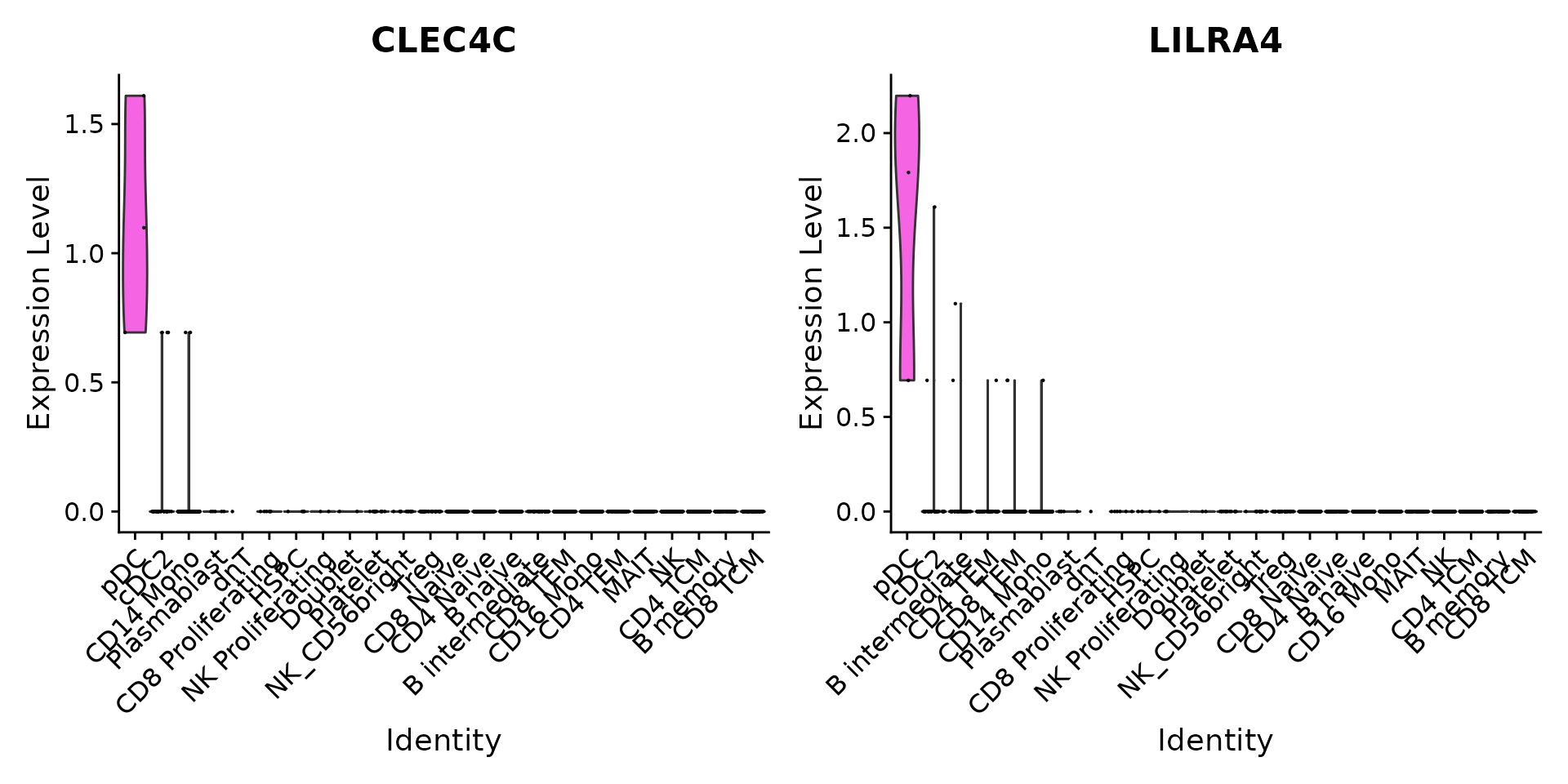
我们可以通过探索典型标记基因的表达来验证我们的预测。例如,CLEC4C 和 LIRA4 已被报道为 pDC 身份的标记,与我们的预测一致。同样,如果我们进行差异表达来识别 Tregs 标记,我们就会识别出一组典型标记,包括 RTKN2、CTLA4、FOXP3 和 IL2RA。
Idents(pbmc3k) <- 'predicted.celltype.l2'
VlnPlot(pbmc3k, features = c("CLEC4C", "LILRA4"), sort = TRUE) + NoLegend()
treg_markers <- FindMarkers(pbmc3k, ident.1 = "Treg", only.pos = TRUE, logfc.threshold = 0.1)
print(head(treg_markers))
## p_val avg_log2FC pct.1 pct.2 p_val_adj
## FOXP3 8.428389e-41 0.2002914 0.152 0.002 1.059617e-36
## RP11-1399P15.1 1.702575e-21 0.4749271 0.273 0.021 2.140478e-17
## RTKN2 3.890305e-17 0.2339929 0.121 0.005 4.890892e-13
## TIGIT 3.773433e-16 0.5934260 0.424 0.065 4.743960e-12
## F5 4.035005e-16 0.2318413 0.121 0.005 5.072808e-12
## FRS2 2.983908e-15 0.1906088 0.152 0.009 3.751369e-11
最后,我们可以可视化表面蛋白的估算水平,这是根据 CITE-seq reference 推断的。
DefaultAssay(pbmc3k) <- 'predicted_ADT'
# see a list of proteins: rownames(pbmc3k)
FeaturePlot(pbmc3k, features = c("CD3-1", "CD45RA", "IgD"), reduction = "ref.umap", cols = c("lightgrey", "darkgreen"), ncol = 3)
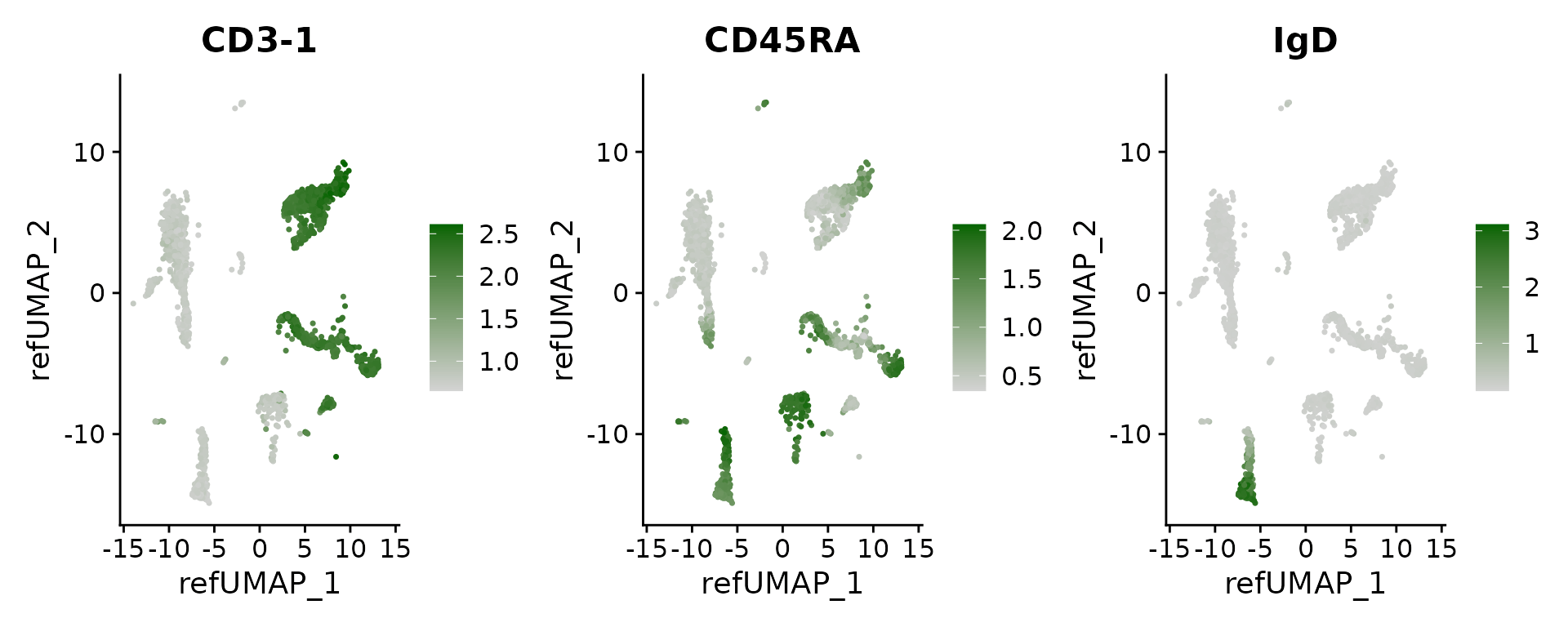
2.4 计算新的 UMAP 可视化
在前面的示例中,我们在映射到 reference-derived UMAP 后可视化 query cells。保持一致的可视化可以帮助解释新数据集。但是,如果 query 数据集中存在未在 reference 中表示的细胞状态,它们将投影到 reference 中最相似的细胞。这是 UMAP 包建立的预期行为和功能,但可能会掩盖 query 中可能感兴趣的新细胞类型的存在。
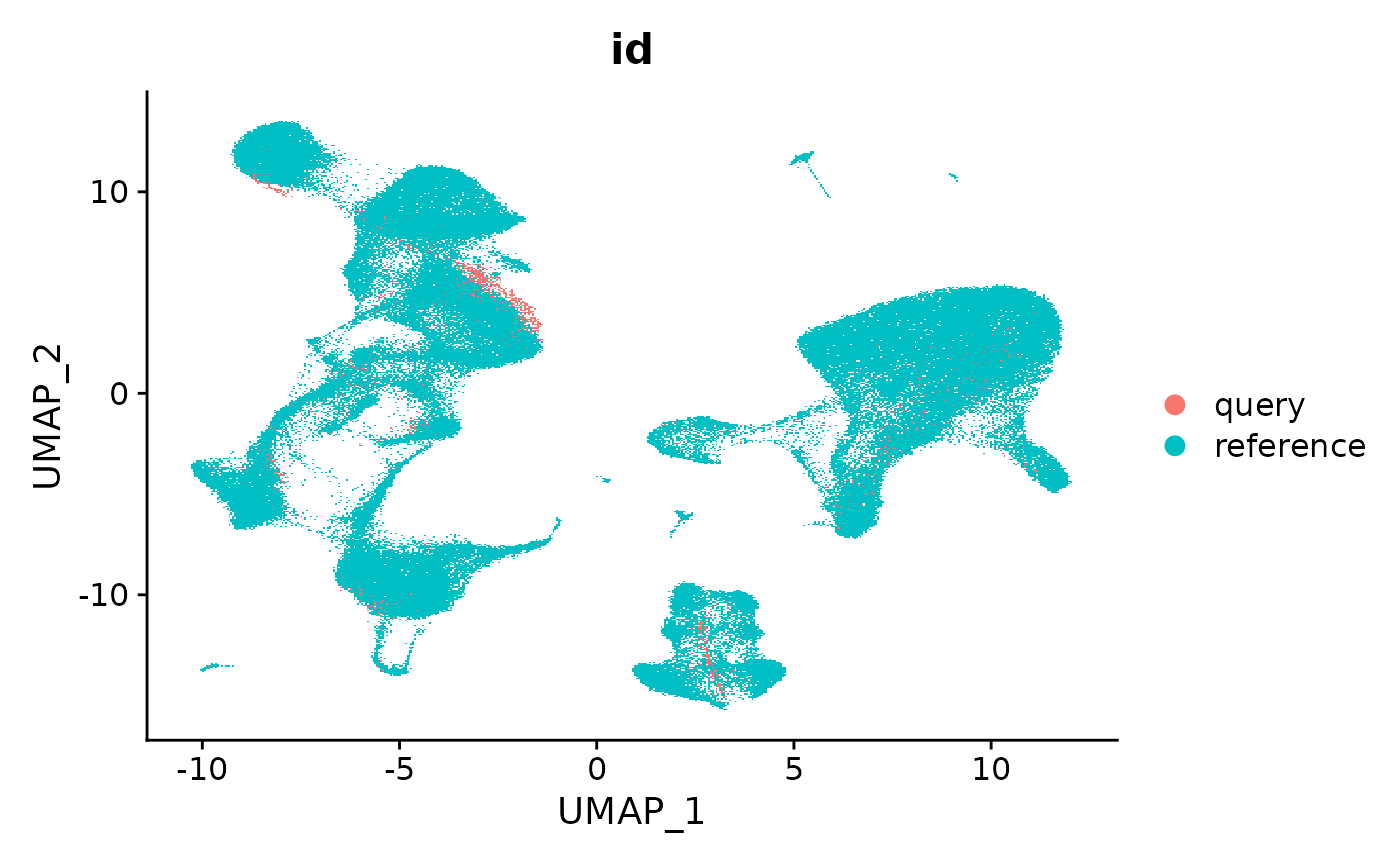
在我们的手稿中,我们映射了一个包含正在发育和分化的中性粒细胞的 query 数据集,这些细胞未包含在我们的 reference 中。我们发现,在合并 reference 和 query 后计算新的 UMAP(“从头可视化”)有助于识别这些群体,如 Supplementary Figure 8 所示。在“从头”可视化中,query 中的独特细胞状态保持分离。在此示例中,2,700 个 PBMC 不包含独特的细胞状态,但我们在下面演示了如何计算此可视化。
我们强调,如果用户尝试映射基础样本不是 PBMC 的数据集,或者包含 reference 中不存在的细胞类型,则计算“从头”可视化是解释其数据集的重要一步。
#merge reference and query
reference$id <- 'reference'
pbmc3k$id <- 'query'
refquery <- merge(reference, pbmc3k)
refquery[["spca"]] <- merge(reference[["spca"]], pbmc3k[["ref.spca"]])
refquery <- RunUMAP(refquery, reduction = 'spca', dims = 1:50)
DimPlot(refquery, group.by = 'id', shuffle = TRUE)
3 示例2:映射人类骨髓细胞图谱
3.1 多模态 BMNC 参考数据集
作为第二个例子,我们绘制了由人类细胞图谱生成的来自八个捐赠者的人类骨髓单核 (BMNC) 细胞的数据集。作为一个 reference,我们使用人类 BMNC 的 CITE-seq reference,并使用加权最近邻分析 (WNN) 对其进行分析。
此小节展示了与上一个栏的 PBMC 示例相同的 reference-mapping 功能。此外,我们还演示:
-
如何构建一个 supervised PCA (sPCA) transformation -
如何将多个数据集连续映射到同一 reference -
进一步提升映射速度的优化步骤
# Both datasets are available through SeuratData
library(SeuratData)
#load reference data
InstallData("bmcite")
bm <- LoadData(ds = "bmcite")
#load query data
InstallData('hcabm40k')
hcabm40k <- LoadData(ds = "hcabm40k")
这一步可能出现报错,推测是因为国外网络的问题,需要手动下载安装
wget http://seurat.nygenome.org/src/contrib/bmcite.SeuratData_0.3.0.tar.gzinstall.packages('bmcite.SeuratData_0.3.0.tar.gz', repos = NULL, type = "source")
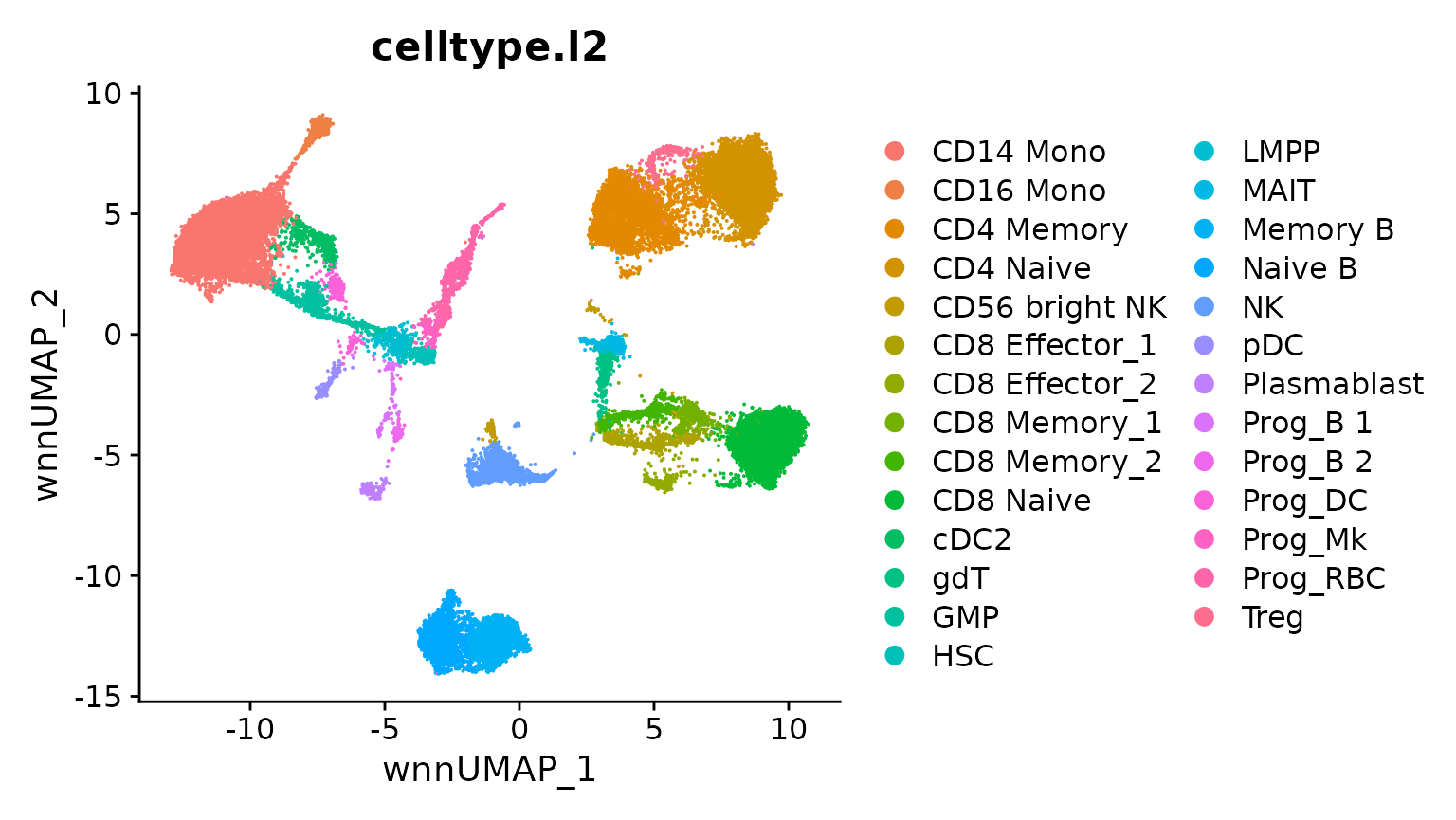
reference 数据集包含一个 WNN graph,反映了此 CITE-seq 实验中 RNA 和蛋白质数据的加权组合。
我们可以根据该图计算 UMAP 可视化。我们设置 return.model = TRUE,这将使我们能够将数据集投影到此可视化上。
bm <- RunUMAP(bm, nn.name = "weighted.nn", reduction.name = "wnn.umap",
reduction.key = "wnnUMAP_", return.model = TRUE)
DimPlot(bm, group.by = "celltype.l2", reduction = "wnn.umap")
3.2 计算 sPCA 变换
正如我们的手稿中所述,我们首先计算 ‘supervised’ PCA。这确定了最能封装 WNN graph 结构的转录组数据的转换。这允许蛋白质和 RNA 测量的加权组合来“监督” PCA,并突出显示最相关的变异来源。计算此转换后,我们可以将其投影到 query 数据集上。我们还可以计算和投影 PCA 投影,但建议在处理通过 WNN 分析构建的多模态 references 时使用 sPCA。
sPCA 计算执行一次,然后可以快速投影到每个 query 数据集上。
bm <- ScaleData(bm, assay = 'RNA')
bm <- RunSPCA(bm, assay = 'RNA', graph = 'wsnn')
3.3 计算缓存的邻居索引
由于我们将多个 query 样本映射到同一 reference,因此我们可以缓存仅涉及该 reference 的特定步骤。此步骤是可选的,但在映射多个样本时会提高速度。
我们计算 reference 的 sPCA 空间中的前 50 个 neighbors。我们将此信息存储在 reference Seurat 对象内的 spca.annoy.neighbors 对象中,并缓存 annoy 索引数据结构(通过 cache.index = TRUE)。
bm <- FindNeighbors(
object = bm,
reduction = "spca",
dims = 1:50,
graph.name = "spca.annoy.neighbors",
k.param = 50,
cache.index = TRUE,
return.neighbor = TRUE,
l2.norm = TRUE
)
如何保存和加载缓存的 annoy 索引?
如果要保存和加载使用 method = "annoy" 和 cache.index = TRUE 生成的 Neighbor 对象的缓存索引,请使用 SaveAnnoyIndex()/LoadAnnoyIndex() 函数。重要的是,该索引无法正常保存到 RDS 或 RDA 文件,因此它不会在 R 会话重新启动或包含它的 Seurat 对象的 saveRDS/readRDS 中正确保留。相反,每次 R 重新启动或从 RDS 加载引用 Seurat 对象时,请使用 LoadAnnoyIndex() 将 Annoy 索引添加到 Neighbor 对象。SaveAnnoyIndex() 创建的文件可以与引用 Seurat 对象一起分发,并添加到 reference 中的 Neighbor 对象中。
bm[["spca.annoy.neighbors"]]
## A Neighbor object containing the 50 nearest neighbors for 30672 cells
SaveAnnoyIndex(object = bm[["spca.annoy.neighbors"]], file = "../data/reftmp.idx")
bm[["spca.annoy.neighbors"]] <- LoadAnnoyIndex(object = bm[["spca.annoy.neighbors"]], file = "../data/reftmp.idx")
3.4 查询数据集预处理
在这里,我们将演示将多个供体骨髓样本映射到多模态骨髓 reference。这些 query 数据集源自人类细胞图谱 (HCA) 免疫细胞图谱骨髓数据集,可通过 SeuratData 获取。该数据集作为具有 8 个捐赠者的单个合并对象提供。我们首先将数据拆分回 8 个独立的 Seurat 对象,每个原始捐赠者分别对应一个对象进行映射。
library(dplyr)
library(SeuratData)
InstallData('hcabm40k')
hcabm40k.batches <- SplitObject(hcabm40k, split.by = "orig.ident")
然后,我们以与 reference 相同的方式归一化 query。此处,通过 NormalizeData() 使用对数标准化对 reference 进行归一化。如果 reference 已使用 SCTransform() 归一化,则 query 也必须使用 SCTransform() 归一化。
hcabm40k.batches <- lapply(X = hcabm40k.batches, FUN = NormalizeData, verbose = FALSE)
3.5 映射
然后,我们在每个捐赠者 query 数据集和多模态 reference 之间找到 anchors。该命令经过优化,通过传入一组预先计算的 reference neighbors 并关闭 anchor 过滤来最大限度地减少映射时间。
anchors <- list()
for (i in 1:length(hcabm40k.batches)) {
anchors[[i]] <- FindTransferAnchors(
reference = bm,
query = hcabm40k.batches[[i]],
k.filter = NA,
reference.reduction = "spca",
reference.neighbors = "spca.annoy.neighbors",
dims = 1:50
)
}
然后我们单独映射每个数据集。
for (i in 1:length(hcabm40k.batches)) {
hcabm40k.batches[[i]] <- MapQuery(
anchorset = anchors[[i]],
query = hcabm40k.batches[[i]],
reference = bm,
refdata = list(
celltype = "celltype.l2",
predicted_ADT = "ADT"),
reference.reduction = "spca",
reduction.model = "wnn.umap"
)
}
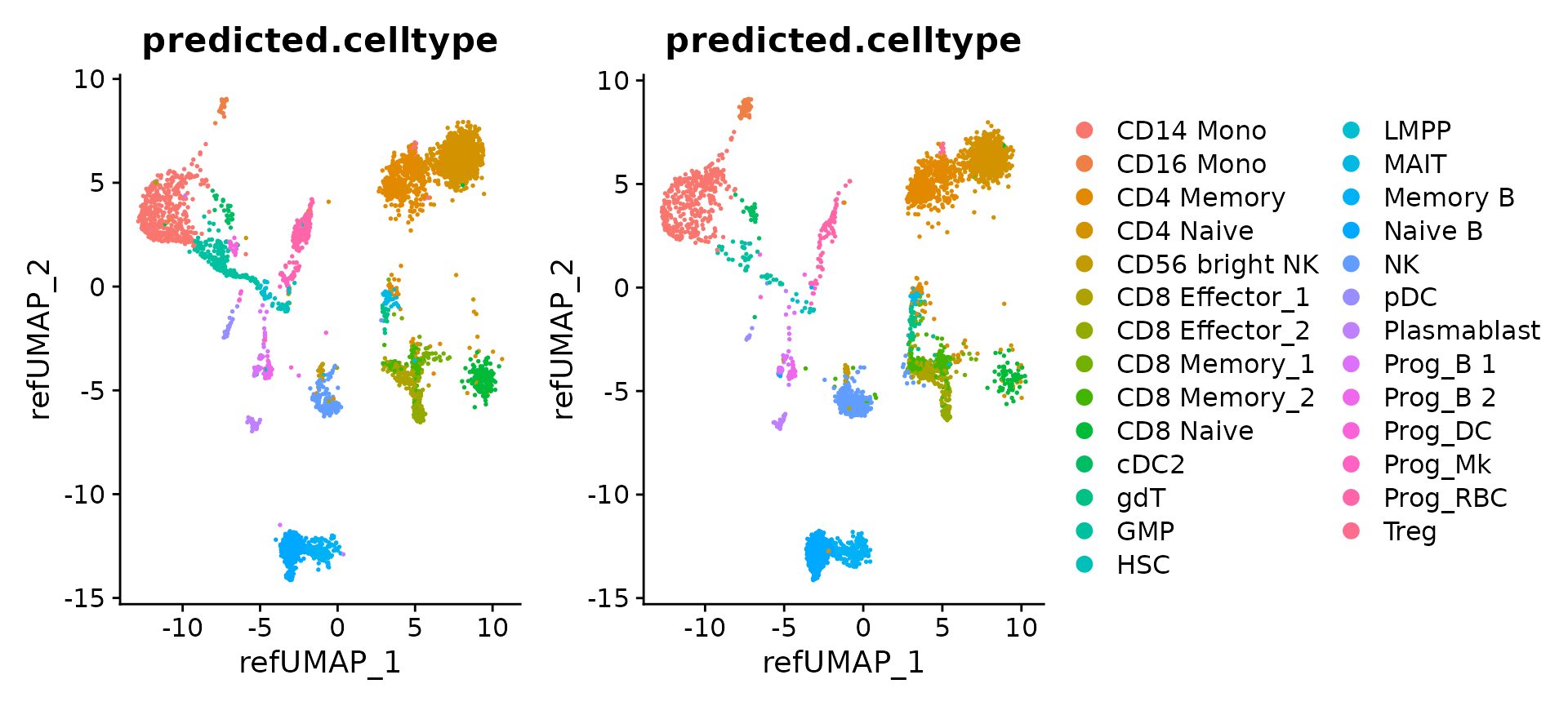
3.6 探索映射结果
现在映射已完成,我们可以可视化各个对象的结果
p1 <- DimPlot(hcabm40k.batches[[1]], reduction = 'ref.umap', group.by = 'predicted.celltype', label.size = 3)
p2 <- DimPlot(hcabm40k.batches[[2]], reduction = 'ref.umap', group.by = 'predicted.celltype', label.size = 3)
p1 + p2 + plot_layout(guides = "collect")
我们还可以将所有对象合并到一个数据集中。请注意,它们都已集成到由 reference 定义的公共空间中。然后我们可以一起可视化结果。
# Merge the batches
hcabm40k <- merge(hcabm40k.batches[[1]], hcabm40k.batches[2:length(hcabm40k.batches)], merge.dr = "ref.umap")
DimPlot(hcabm40k, reduction = "ref.umap", group.by = "predicted.celltype", label = TRUE, repel = TRUE, label.size = 3) + NoLegend()
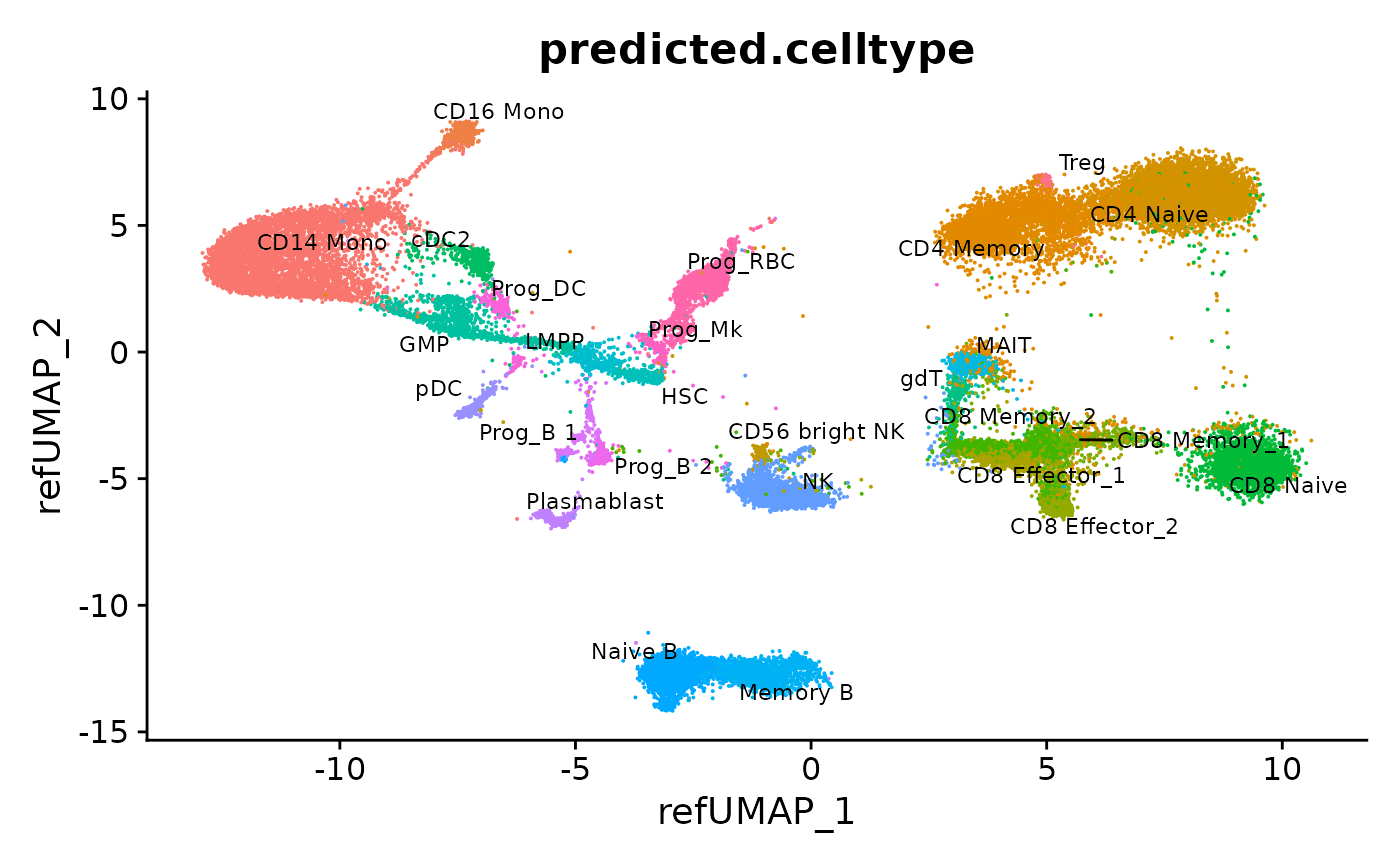
我们可以可视化 query cells 中的基因表达、聚类预测分数和(估算的)表面蛋白水平:
p3 <- FeaturePlot(hcabm40k, features = c("rna_TRDC", "rna_MPO", "rna_AVP"), reduction = 'ref.umap',
max.cutoff = 3, ncol = 3)
# cell type prediction scores
DefaultAssay(hcabm40k) <- 'prediction.score.celltype'
p4 <- FeaturePlot(hcabm40k, features = c("CD16 Mono", "HSC", "Prog-RBC"), ncol = 3,
cols = c("lightgrey", "darkred"))
# imputed protein levels
DefaultAssay(hcabm40k) <- 'predicted_ADT'
p5 <- FeaturePlot(hcabm40k, features = c("CD45RA", "CD16", "CD161"), reduction = 'ref.umap',
min.cutoff = 'q10', max.cutoff = 'q99', cols = c("lightgrey", "darkgreen") ,
ncol = 3)
p3 / p4 / p5

本文由 mdnice 多平台发布















 3187
3187











 被折叠的 条评论
为什么被折叠?
被折叠的 条评论
为什么被折叠?
 到【灌水乐园】发言
到【灌水乐园】发言









