






 文章介绍了两种生存分析平台——GEPIA和Cbioportal的用途,并分享了如何从UCSCXena下载数据进行生存分析。通过R代码,文章详细阐述了整合临床和表达数据、建立Cox模型以及确定最佳cutoff值来绘制生存曲线的过程。
文章介绍了两种生存分析平台——GEPIA和Cbioportal的用途,并分享了如何从UCSCXena下载数据进行生存分析。通过R代码,文章详细阐述了整合临床和表达数据、建立Cox模型以及确定最佳cutoff值来绘制生存曲线的过程。
一、生存分析的平台(直接使用平台做生存分析)
生存分析的平台有很多,我使用过以下两种,感觉比较稳定也不错:
- GEPIA:适合生成生存曲线可视化结果(可以作为参考,但我倾向于自己抽取数据写R代码来画生存曲线)
- Cbioportal:可以用来下载临床生存数据,也可以可视化
平台的使用教程就不在此处赘述了,相信大家都可以搜到
二、自己写代码进行生存分析(方便可视化图的调整和修改)
数据来源
做生存分析需要两类数据作为输入:
- 临床生存数据
- 表达量数据
注意:二者的样本要对应
数据的来源有很多,我目前使用过从UCSC Xena下载数据做生存分析,简述一下如何下载数据和生成生存曲线
从UCSC Xena下载数据
从博主的UCSCXenaTools学到的。
1.临床数据

点开之后点击download下载即可,得到txt文件,对于生存数据的相关指标的描述在另一名博主的教程里做了明确的解释:
OS/PFS/PFI/DFS/DFI/DSS各种生存指标定义
2.表达数据
因为要选择和临床生存数据所匹配的表达数据,所以也要从相应的平台下载(sample要对应嘛)
根据UCSCXenaTools的教程选择第二种(虽然我也不知道为什么啦,知道原因的欢迎评论区留言哦)
点开之后点击download下载即可,得到压缩包,里面文件可以用txt打开
生存曲线
1.首先要整合临床数据和表达数据,按照UCSCXenaTools的教程,整理成如图所示的格式(注意表达量和时间、状态都是数字类型哦)
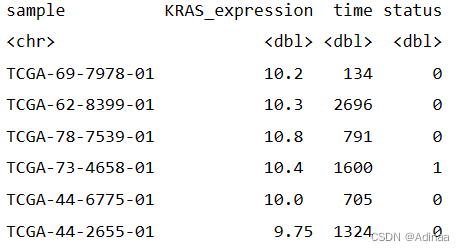
表达量可以有多个基因的多列,只要样本、time、status对应无误即可,例如:
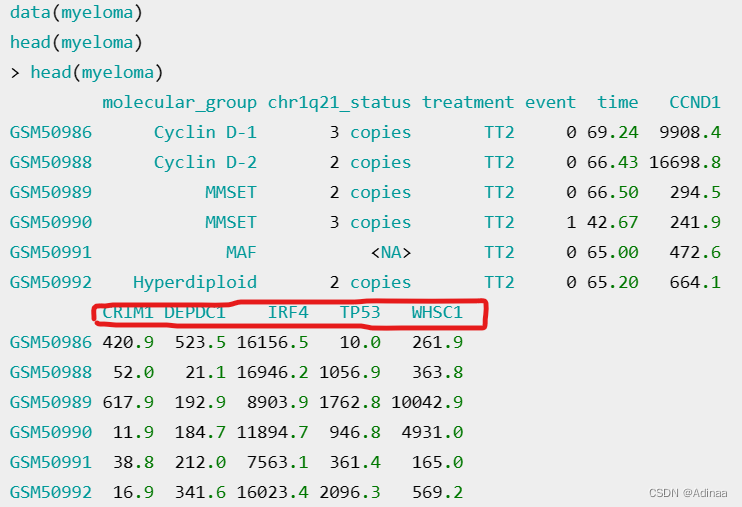
2.整理好数据之后,就按照Survival analysis的流程做起
首先导入包
library(survival)
library(survminer)
之后Cox model
merged_data就是之前整合好的数据,KRAS_expression,time, status都是merged_data的列名,分别代表要分析的基因的表达量、生存时间、生存状态
fit = coxph(Surv(time, status) ~ KRAS_expression, data = merged_data)
之后取最佳cutoff(截断)【很重要,不然pvalue不能<0.05(显著)】
按照博主的流程来即可取最佳cutoff(截断)并绘制生存曲线
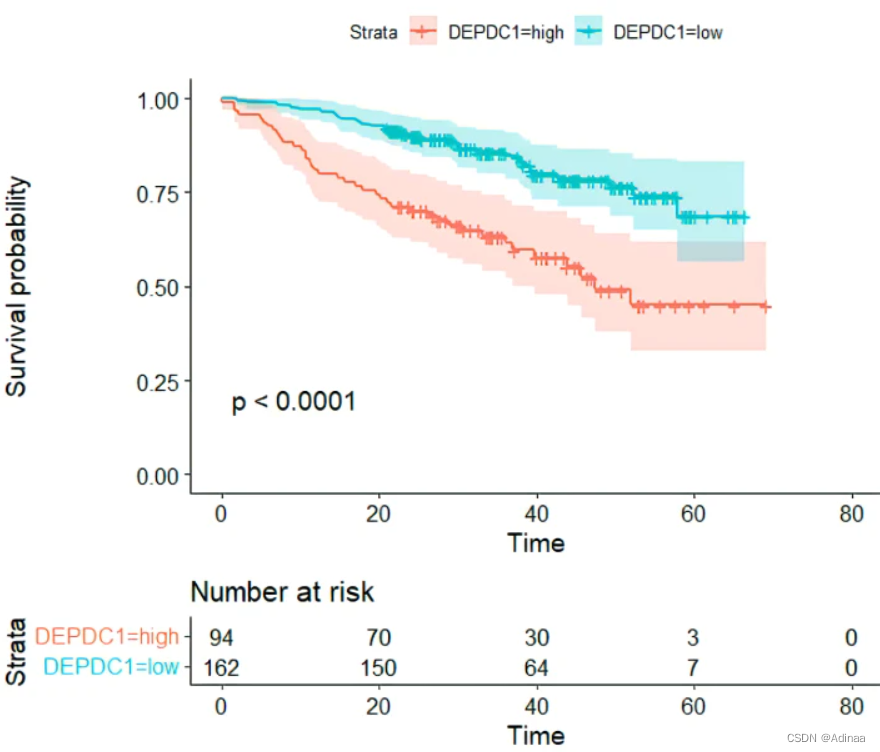
还有具体绘制的详细参数设置教程生存曲线图详细绘图教程
补充:若要修改风险表等表的具体参数可在ggsurvplot内添加tables.theme,例如
tables.theme=theme(
plot.title=element_text(face="bold",size=15),
axis.title.x = element_text(size=20,face="bold",family="Arial" ),
axis.text.y=element_text(size=13,face="bold"),
axis.text.x=element_text(size=15,color="black"))
再次感谢提供教程的博主们,此处整理供大家方便汲取博主们的经验

















 996
996

 被折叠的 条评论
为什么被折叠?
被折叠的 条评论
为什么被折叠?
 到【灌水乐园】发言
到【灌水乐园】发言









