






 本文介绍了在生物信息学中,如何通过SAM文件中的标签判断reads的唯一性(Uniquereads)、错配情况以及duplication的识别。讨论了保留或过滤这些类型reads的决策依据,如测序深度、产品质量和计算方法。
本文介绍了在生物信息学中,如何通过SAM文件中的标签判断reads的唯一性(Uniquereads)、错配情况以及duplication的识别。讨论了保留或过滤这些类型reads的决策依据,如测序深度、产品质量和计算方法。
序列比对
比对情况
Unique reads : 仅有一个最优比对位置的reads,因此有多个最优比对位置的reads就不是unique reads
- 如何判断一条reads是否是unique reads?
1.sam文件中XT:A:{} 标签,其中U表示仅有一个最优比对,R表示有多个最优比对


2.sam文件中X0:1:n 标签,其中n表示共有多少个最优比对
![[图片]](https://i-blog.csdnimg.cn/blog_migrate/caf2f4f1299b56ef0b296cb0e410588d.png)
错配: bwa中可以指定-n 参数指定最大允许的错配数量
- 如何查看序列的错配信息:sam文件的MD:Z:标签
duplication : 由于pcr偏好性导致pcr扩增中出现多条重复reads,测序结果表明多条reads比对到同一个位置
- 根据泊松定理可以简单的知道,在实际情况下比对到同一位置的reads数量应该非常少,所以duplication主要由于实验误差产生
reads保留
是否应该保留duplication reads?
- Duplication reads 通常表示对于同一段序列的多次拷贝,往往是由于实验误差导致的,对于揭示生物信息没有意义,一般保留过滤唯一值即可。
- Duplication reads过多表示测序质量不高,二代测序的产品质量要求重复率< 10%
重复率计算公式:(1-Duplication reads)/raw reads
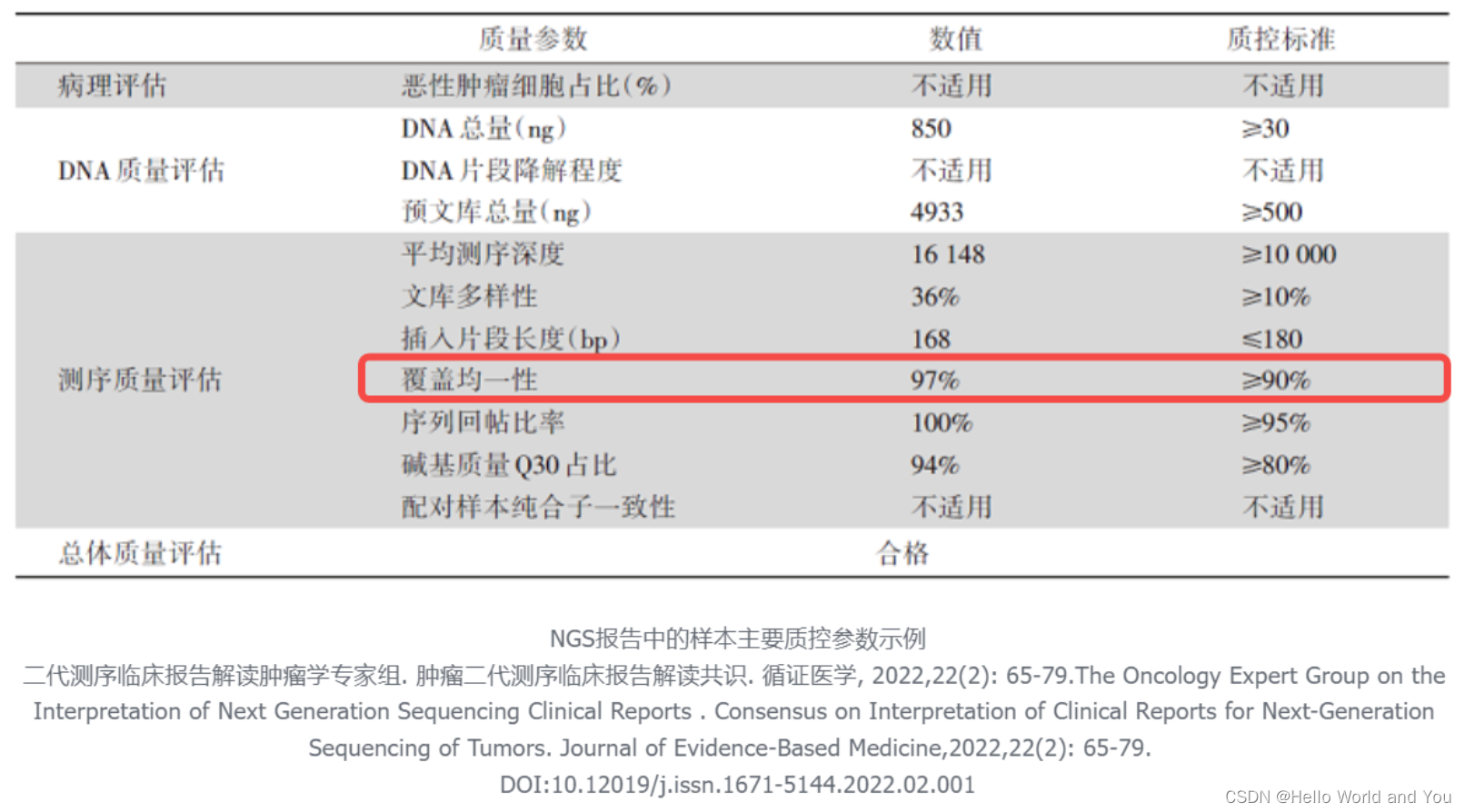
是否应该保留有错配/非Unique reads?
- 需要根据测序深度,计算方法,产品要求而定,不能一概而论。
- 目前基本的测试结果和文献证据表明测序数据量不足,结果假阳率明显增且高灵敏度下降。保留部分错配/非Unique reads收益大于不保留。

















 2898
2898

 被折叠的 条评论
为什么被折叠?
被折叠的 条评论
为什么被折叠?
 到【灌水乐园】发言
到【灌水乐园】发言









