






相信各位做单细胞的童鞋们,都遇到过去除多细胞的情况,而Cellranger默认的是前1%(3000cell),对于低质量的细胞有了算法来确认是否保留,但是对于多细胞而言,Cellranger和Seurat并没有相应的算法进行去除,今天给大家介绍一个去除多细胞的方法-DoubletFinder,一个R包,现在我们开回顾一下这个包的用法
DoubletFinder概述
DoubletFinder工作流程是从处理过的Seurat对象开始。
seu_kidney <- CreateSeuratObject(kidney.data)
seu_kidney <- NormalizeData(seu_kidney)
seu_kidney <- ScaleData(seu_kidney)
seu_kidney <- FindVariableFeatures(seu_kidney, selection.method = "vst", nfeatures = 2000)
seu_kidney <- RunPCA(seu_kidney)
seu_kidney <- RunUMAP(seu_kidney, dims = 1:10)
## Pre-process Seurat object (sctransform) -----------------------------------------------------------------------------------
seu_kidney <- CreateSeuratObject(kidney.data)
seu_kidney <- SCTransform(seu_kidney)
seu_kidney <- RunPCA(seu_kidney)
seu_kidney <- RunUMAP(seu_kidney, dims = 1:10)
至于更多的Seurat的细节处理,这里不再详述,大家可以根据自己的需求编写Seurat代码,我们这里从构建好Seurat对象开始分析DoubletFinder。
然后,通过对通过随机抽样与替换选择的细胞对的基因表达谱求平均,从原始UMI计数矩阵中生成Artificial doublets。然后生成足够的Artificial doublets,占合成数据的25%(pN = 0.25)。接下来,使用原始数据分析工作流程中使用的相同scale,缩放和可变基因定义参数,对真实数据和人工数据进行合并和预处理。值得注意的是,为了保留singlets and doublets之间的差异,在合并的人工数据集预处理期间不执行nUMI回归。使用在原始数据预处理期间选择的具有统计意义的相同数量的PC,然后使用“ fields” R包中的“ rdist”函数将PC单元嵌入转换为欧几里得距离矩阵(Nychka等人,2017)。
然后,从此距离矩阵中定义每个单元格的最近邻居,然后通过将每个实际单元格的人工邻居数除以邻域大小(pK),为每个实际单元格计算人工最近邻居的比例。然后,将最终的doublet分类分配给具有最高pANN的单元格,其中将n设置为具有或不具有同型doublet调整的期望doublet的总数。
在这里我们只用单细胞数据来进行模拟
DoubletFinder可以分为4个步骤:
一、 从现有的scRNA-seq数据生成 artificial doublets
二、预处理合并的real-artificial data
三、执行PCA并使用PC距离矩阵来找到每个单元的人工k最近邻(pANN)的比例
四、根据 expected number of doublets排列顺序和阈值pANN值
DoubletFinder采用以下参数:
(1)seu〜这是一个经过充分处理的Seurat对象( after NormalizeData, FindVariableGenes, ScaleData, RunPCA, and RunTSNE have all been run)
(2)PC〜具有统计意义的主成分数,指定为范围(e.g., PCs = 1:10)
(3)pN〜定义生成的人工双峰的数量,表示为合并的真实人工数据的一部分。 基于DoubletFinder性能在很大程度上是pN是不变的,默认设置为25%
(4)pK〜定义用于计算pANN的PC邻域大小,表示为合并的真实real-artificial数据的一部分。 未设置默认值,因为应该为每个scRNA-seq数据集调整pK。 最佳pK值应使用以下描述的策略进行估算。
(5)nExp〜这定义了用于进行最终doublet/singlet预测的pANN阈值。 可以从10X / Drop-Seq装置中的细胞装载密度来最好地估计该值,并根据homotypic doublets的估计比例进行调整。
'Best-Practices' for scRNA-seq data generated without sample multiplexing
Input scRNA-seq Data
不要将DoubletFinder应用于代表多个不同样本的汇总scRNA-seq数据。 例如,如果您对表示跨10Xlane测序的WT和突变细胞系的汇总数据运行DoubletFinder,则将从WT和突变细胞生成artificial doublet,它们在您的数据中不存在。 这些artificial doublets 会歪曲结果。 值得注意的是,可以对通过在多个10Xlane上分割单个样本而生成的数据运行DoubletFinder。
确保清除输入数据中的低质量cluster
pK Selection
通过对模拟的scRNAseq数据进行pN-pK参数扫描的ROC分析,其中(I)细胞状态数目可变,并且(II)转录异质性的变量大小表明(I)最佳pK值选择取决于细胞状态总数,并且(II )DoubletFinder性能在应用于转录同源数据时会受到影响。
使用BCMVN最大化优化pK
双峰系数(BC)测量数据分布中与单峰的偏差.对于DoubletFinder参数拟合,我们认为产生非单峰pANN分布的参数集将最佳地将单重态与双态分离,结果将表现最佳。 Thus scRNA-seq datasets,我们测试了在pN-pK参数扫描期间生成的每个pANN分布,以发现具有较高BC值的pANN分布。 具体来说,我们按照modes R包中的“ bimodality_coefficient”函数中实现的方式计算了BC,其形式为:
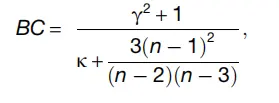
$9W7KS9~PDTGZ8ZUG}J3E9D.png
其中g是pANN分布偏度,k是峰度,n是样本大小。 然后,我们测量了在所有测试的pN值中每个pK的BC平均值和方差,因为先前已证明DoubletFinder的性能不受生成的人工doublet数量的影响。
我们记录了此工作流程的结果。当pK值太高(例如pK> 0.1)时, singlets and doublets 具有相似的人工最近邻比例,并且所得的pANN分布与低BC和AUC有关。相反,当pK值太低(例如pK = 5e-4)时,DoubletFinder性能会受到影响,因为基因表达空间中的邻域受到导致多峰pANN分布的局部效应的支配。由于这些分布不是单峰分布,因此它们与高BC相关。
但是,局部效应对集成到数据集中(pN)的人工双峰的数量敏感,导致相关pK值的BC方差升高。 最后,理想的pK值(例如pK = 0.01)会生成长尾pANN分布,其特征是高AUC和高BC,且方差低。 我们利用这些观察结果设计了一个新的pK参数选择指标-BCMVN-形式为;
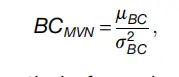
2.png
分别是pN值中每个pK的BC平均值和方差。 这项研究中测试的四个数据集的BCMVN分布具有一个单一的,视觉上可分辨的最大值。 对于真实数据集,此最大值与通过ROC分析确定的理想pK值相对应。
sweep.res.list_kidney <- paramSweep_v3(pbmc, PCs = 1:20, sct = FALSE)
#head(sweep.res.list_kidney)
sweep.stats_kidney <- summarizeSweep(sweep.res.list_kidney, GT = FALSE)
#head(sweep.res.list_kidney)
bcmvn_kidney <- find.pK(sweep.stats_kidney)
根据上面的描述,最佳的pk值应该是
mpK<-as.numeric(as.vector(bcmvn_kidney$pK[which.max(bcmvn_kidney$BCmetric)]))
annotations <- pbmc@meta.data$seurat_clusters
## Homotypic Doublet Proportion Estimate -------------------------------------------------------------------------------------
homotypic.prop <- modelHomotypic(annotations) ## ex: annotations <- seu_kidney@meta.data$ClusteringResults
nExp_poi <- round(0.075*length(seu_kidney@cell.names)) ## Assuming 7.5% doublet formation rate - tailor for your dataset
nExp_poi.adj <- round(nExp_poi*(1-homotypic.prop))
最后,我们运用
pbmc <- doubletFinder_v3(pbmc, PCs = 1:20, pN = 0.25, pK = mpK, nExp = nExp_poi, reuse.pANN = FALSE, sct = FALSE)
就大功告成了~~~~~

















 5565
5565

 被折叠的 条评论
为什么被折叠?
被折叠的 条评论
为什么被折叠?
 到【灌水乐园】发言
到【灌水乐园】发言









