







TCGA数据已经改版,workflow.type只有STAR-counts数据,改版后数据下载与预处理方法见:
TCGA测序数据改版后的应对_老实人谢耳朵的博客-CSDN博客
以下为改版前的方法:
library("TCGAbiolinks")
library("SummarizedExperiment")一、数据下载
1 GDCquery筛选-下载数据数据
query <- GDCquery(project = "TCGA-PRAD", #项目名
data.category = "Transcriptome Profiling",
data.type = "Gene Expression Quantification",
workflow.type = "HTSeq - Counts")
samplesDown <- getResults(query,cols=c("cases"))
#从sampleDown检索出TP
dataSmTP <- TCGAquery_SampleTypes(barcode = samplesDown,
typesample = "TP") #组织类型
#筛选完成的query
queryDown <- GDCquery(project = "TCGA-PRAD",
data.category = "Transcriptome Profiling",
data.type = "Gene Expression Quantification",
workflow.type = "HTSeq - Counts",
barcode = dataSmTP)
#下载GDCquery的结果
GDCdownload(queryDown,
method = "api",
directory = "GDCdata",
files.per.chunk = 6) #网速慢可以设小一点二、数据预处理
1 读入数据、预处理
dataPrep1 <- GDCprepare(query = queryDown,
save = TRUE,
directory = 'GDCdata', #默认
save.filename ="PRAD_case.rda")
dataPrep2 <- TCGAanalyze_Preprocessing(object = dataPrep1,
cor.cut = 0.6,
datatype = "HTSeq - Counts")
purityDATA <- TCGAtumor_purity(colnames(dataPrep1), 0, 0, 0, 0, 0.6)
Purity.barcodes<-purityDATA$pure_barcodes #肿瘤样本barcodes,为“character”
normal.barcodes<-purityDATA$filtered #正常组织的数据barcodes,为“character”
puried_data <-dataPrep2[,Purity.barcodes] #筛选后数据TCGAtumor_purity():筛选得到一个list[2]。
其中 pure_barcodes 为肿瘤样本,filtered 为正常组织样本(如果GDCquery时下载了Norm数据)
2 基因注释、预处理
需要注意DESeq2包要求数据是未经过标准化的
rownames(puried_data)<-rowData(dataPrep1)$external_gene_name
#dataNorm <- TCGAanalyze_Normalization(tabDF = puried_data,
geneInfo = geneInfo,
method = "gcContent")
dataFilt <- TCGAanalyze_Filtering(tabDF = dataNorm,
method = "quantile",
qnt.cut = 0.25) #过滤阈值
write.csv(dataFilt,file = "dataFilt.csv",quote = FALSE) TCGAbiolinks包下载
该包可以从 Bioconductor 上安装稳定版本
if (!requireNamespace("BiocManager", quietly=TRUE))
install.packages("BiocManager")
BiocManager::install("TCGAbiolinks")或者从 GitHub 上安装开发版本
if (!requireNamespace("BiocManager", quietly = TRUE))
install.packages("BiocManager")
BiocManager::install("BioinformaticsFMRP/TCGAbiolinksGUI.data")
BiocManager::install("BioinformaticsFMRP/TCGAbiolinks")GDCquery参数说明:
TCGA数据下载—TCGAbiolinks包参数详解 - 组学大讲堂问答社区
1.project
获取TCGA中最新的不同癌种的项目号,去官网上找:
“Search”或者从“Program”中选择找

2.data.category
可以指定project中的数据类型,如查询"TCGA-LUAD",有7种数据类型。case_count为病人数,file_count为对应的文件数。下载表达谱,可以设置data.category="Transcriptome Profiling":
> TCGAbiolinks:::getProjectSummary("TCGA-LUAD") #查看指定project的数据类型
$data_categories
case_count file_count data_category
1 80 397 Transcriptome Profiling
2 92 361 Copy Number Variation
3 92 744 Simple Nucleotide Variation
4 80 80 DNA Methylation
5 92 105 Clinical
6 92 352 Sequencing Reads
7 92 517 Biospecimen
$case_count
[1] 92
$file_count
[1] 2556
$file_size
[1] 3.920606e+123.data.type
这个参数受到上一个参数的影响,不同的data.category,会有不同的data.type,如下表所示:

4.workflow.type
这个参数受到上两个参数的影响,不同的data.category和不同的data.type,会有不同的workflow.type,如下表所示:

5. barcode
指定要下载的样品,例如:
barcode =c"TCGA-14-0736-02A-01R-2005-01""TCGA-06-0211-02A-02R-2005-01"6. sample.type
对样本的类型进行过滤,例如,原发癌组织,复发癌等等;

例如:
query <- GDCquery(project = "TCGA-ACC",
data.category = "Transcriptome Profiling",
data.type = "Gene Expression Quantification",
workflow.type = "HTSeq - Counts",
sample.type = c("Primary solid Tumor"),
barcode = c("TARGET-20-PADZCG-04A-01R","TARGET-20-PARJCR-09A-01R"))
TCGA条码(barcode):
1.概念:条码(barcode)是TCGA纳入的每一个标本的专有标识符。
2.结构:
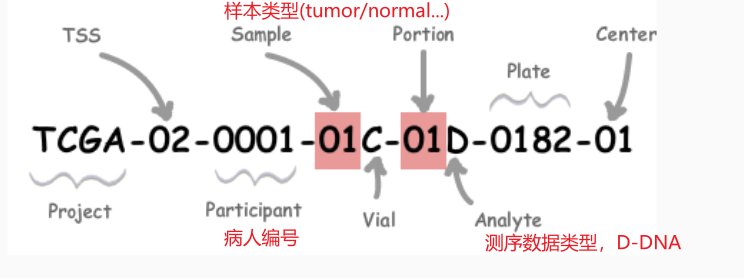
根据前3项即可确定病人编号,如TCGA-02-0001;
其中比较重要的是交代样本类型的Sample的两位数信息,是后面进行差异分析的分组依据。具体对应的含义如下:

3.barcode编制过程如下:
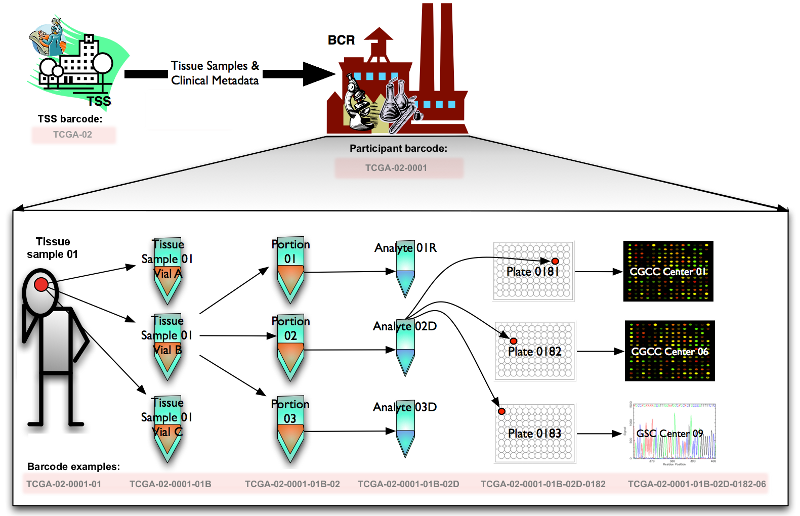
















 1897
1897











 被折叠的 条评论
为什么被折叠?
被折叠的 条评论
为什么被折叠?
 到【灌水乐园】发言
到【灌水乐园】发言











