







 本文介绍了如何使用R包easyTCGA从TCGA数据库下载mRNA、lncRNA、临床信息等数据,并进行了差异分析、生存分析、突变分析和基因表达比较。涉及的R包包括TCGAbiolinks、DESeq2、edgeR和maftools。
本文介绍了如何使用R包easyTCGA从TCGA数据库下载mRNA、lncRNA、临床信息等数据,并进行了差异分析、生存分析、突变分析和基因表达比较。涉及的R包包括TCGAbiolinks、DESeq2、edgeR和maftools。
#使用easyTCGA获取数据
#清空
rm(list=ls())
gc()
# 安装bioconductor上面的R包
options(BioC_mirror="https://mirrors.tuna.tsinghua.edu.cn/bioconductor")
if(!require("BiocManager")) install.packages("BiocManager")
if(!require("TCGAbiolinks")) BiocManager::install("TCGAbiolinks")
if(!require("SummarizedExperiment")) BiocManager::install("SummarizedExperiment")
if(!require("DESeq2")) BiocManager::install("DESeq2")
if(!require("edgeR")) BiocManager::install("edgeR")
if(!require("limma")) BiocManager::install("limma")
# 安装cran上面的R包
if(!require("survival")) install.packages("survival")
if(!require("broom")) install.packages("broom")
if(!require("devtools")) install.packages("devtools")
if(!require("cli")) install.packages("cli")
#devtools::install_github("ayueme/easyTCGA")
library(easyTCGA)
help(package="easyTCGA")
setwd("F:\\TCGA\\TCGA-COAD")
#下载mRNA、lncRNA和临床信息
COAD<-getmrnaexpr("TCGA-COAD")#原始下载的count, TPM, FPKM 均没有经过log2转化
#下载miRNA
COAD_miRNA<-getmirnaexpr("TCGA-COAD")
#下载copy number variation data
COAD_cnv<-getcnv("TCGA-COAD")
#下载masked somatic mutation 体细胞突变
COAD_snv<-getsnvmaf("TCGA-COAD")
#下载DNA methylation beta value 甲基化数据
getmethybeta("TCGA-COAD")
#从下载目录中打开数据
#差异分析
diff<-diff_analysis(exprset=mrna_expr_counts,#没有经过log2转化
project="TCGA-COAD",
save=F)
#批量生存分析
surv<-batch_survival(
exprset=mrna_expr_counts,
clin=clin_info,
is_count = T,
optimal_cut = TRUE,
project="TCGA-COAD",
save_data = FALSE,
min_sample_size = 5,
print_index = TRUE
)
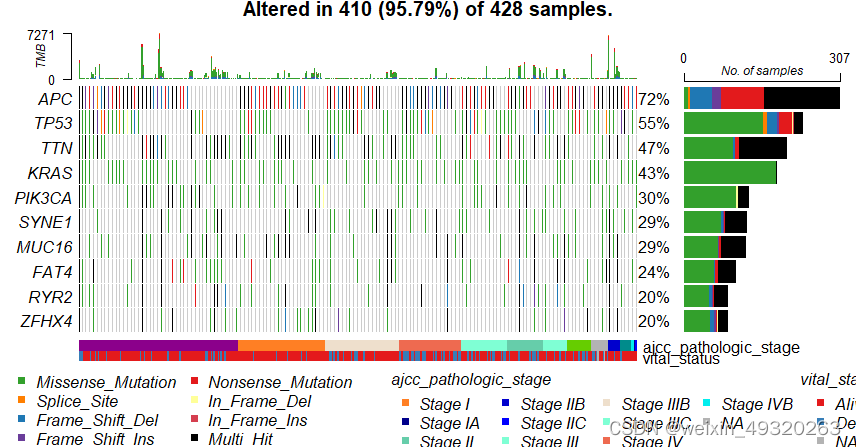
#突变分析:瀑布图
#BiocManager::install("maftools")
library(maftools)
maf<-read.maf(snv,clinicalData=clin_snv)
plotmafSummary(maf)
colnames(clin_snv)
oncoplot(maf=maf,
clinicalFeatures=c("ajcc_pathologic_stage","vital_status"),
top=10,
sortByAnnotation=T
)

#绘制KM曲线
dim(mrna_expr_counts)
set.seed(123)
colnames(clin_info)
clin<-data.frame(time=clin_info$days_to_last_follow_up,
event=clin_info$vital_status)
clin$event<-ifelse(clin$event=="Alive",0,1)
plot_KM(exprset=mrna_expr_counts,
marker="CHPF", #基因
clin=clin,
optimal_cut = TRUE,
return_data = TRUE)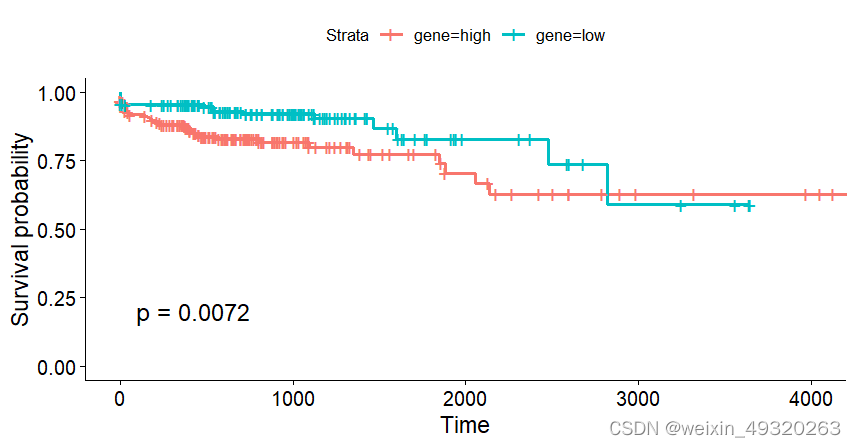
#正常和癌症组织基因表达对比箱线图
rownames(mrna_expr_counts)
plot_gene_paired(exprset=mrna_expr_counts,
marker="CHPF", #基因
return_data = TRUE)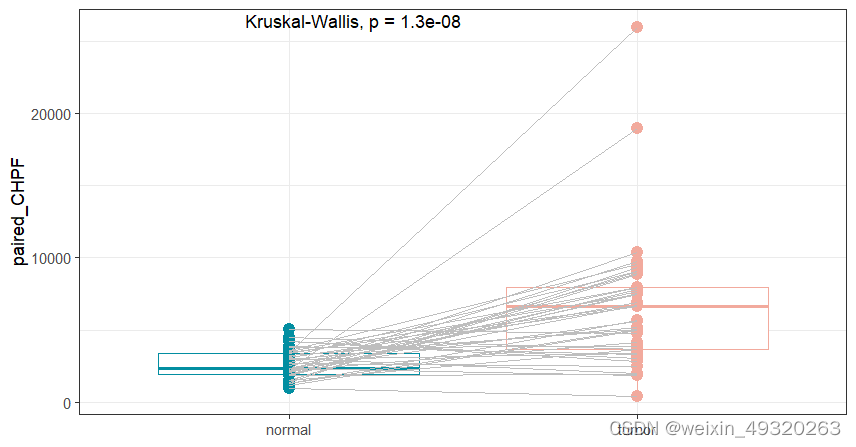
#比较组间基因表达差异
set.seed(123)
group=sample(c(0,1),524,replace = T)
plot_gene(exprset=mrna_expr_counts,
marker=c("CHPF","MAOA"),
group=group,
return_data = TRUE)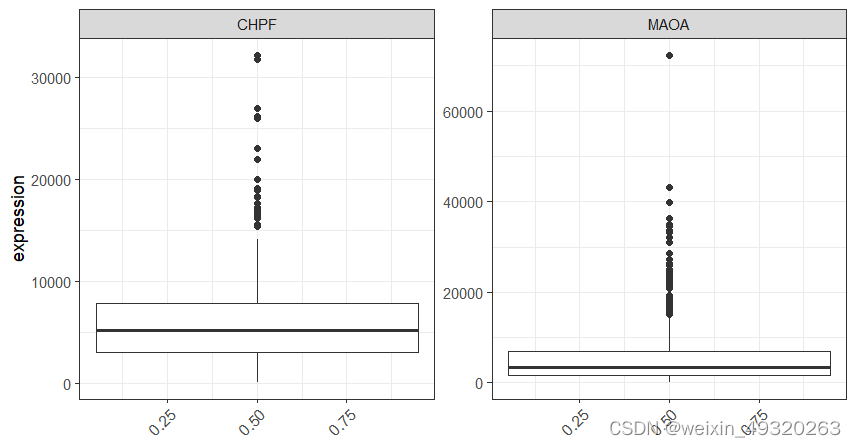


















 1609
1609

 被折叠的 条评论
为什么被折叠?
被折叠的 条评论
为什么被折叠?
 到【灌水乐园】发言
到【灌水乐园】发言









