







 这篇博客详细记录了如何使用R和TCGAbiolinks包从TCGA获取STAR-counts转录组数据,包括下载、整合tsv和json文件,以及数据预处理步骤,如提取count矩阵、转换数据类型和基因注释。过程中遇到了TCGAbiolinks包基因注释失败的问题,但最终通过手动处理得到了预处理后的数据矩阵。
这篇博客详细记录了如何使用R和TCGAbiolinks包从TCGA获取STAR-counts转录组数据,包括下载、整合tsv和json文件,以及数据预处理步骤,如提取count矩阵、转换数据类型和基因注释。过程中遇到了TCGAbiolinks包基因注释失败的问题,但最终通过手动处理得到了预处理后的数据矩阵。
TCGA改版后,workflow.type只有STAR-counts数据,先对所尝试的几种处理方法进行记录:
R version 4.1.2 ; TCGAbiolinks version 2.23.11
方法1
最新版TCGA 矩阵整理,百分百复现成功_sayhello1025的博客-CSDN博客
一、从TCGA网站上下载tsv文件和json文件
1 tsv文件
query <- GDCquery(project = "TCGA-PRAD", #项目名
data.category = "Transcriptome Profiling",
data.type = "Gene Expression Quantification",
workflow.type = "STAR - Counts")
t_samplesDown <- getResults(query,cols=c("cases"))
#从sampleDown检索出TP
t_dataSmTP <- TCGAquery_SampleTypes(barcode = t_samplesDown,
typesample = "TP") #可选
#筛选完成的query
queryDown <- GDCquery(project = "TCGA-PRAD",
data.category = "Transcriptome Profiling",
data.type = "Gene Expression Quantification",
workflow.type = "STAR - Counts",
barcode = t_dataSmTP)
#下载GDCquery的结果
GDCdownload(queryDown,
method = "api",
directory = "GDCdata_star_count",
files.per.chunk = 6) #网速慢可以设小一点跟如下从download 中下载cart效果一样
2 json文件
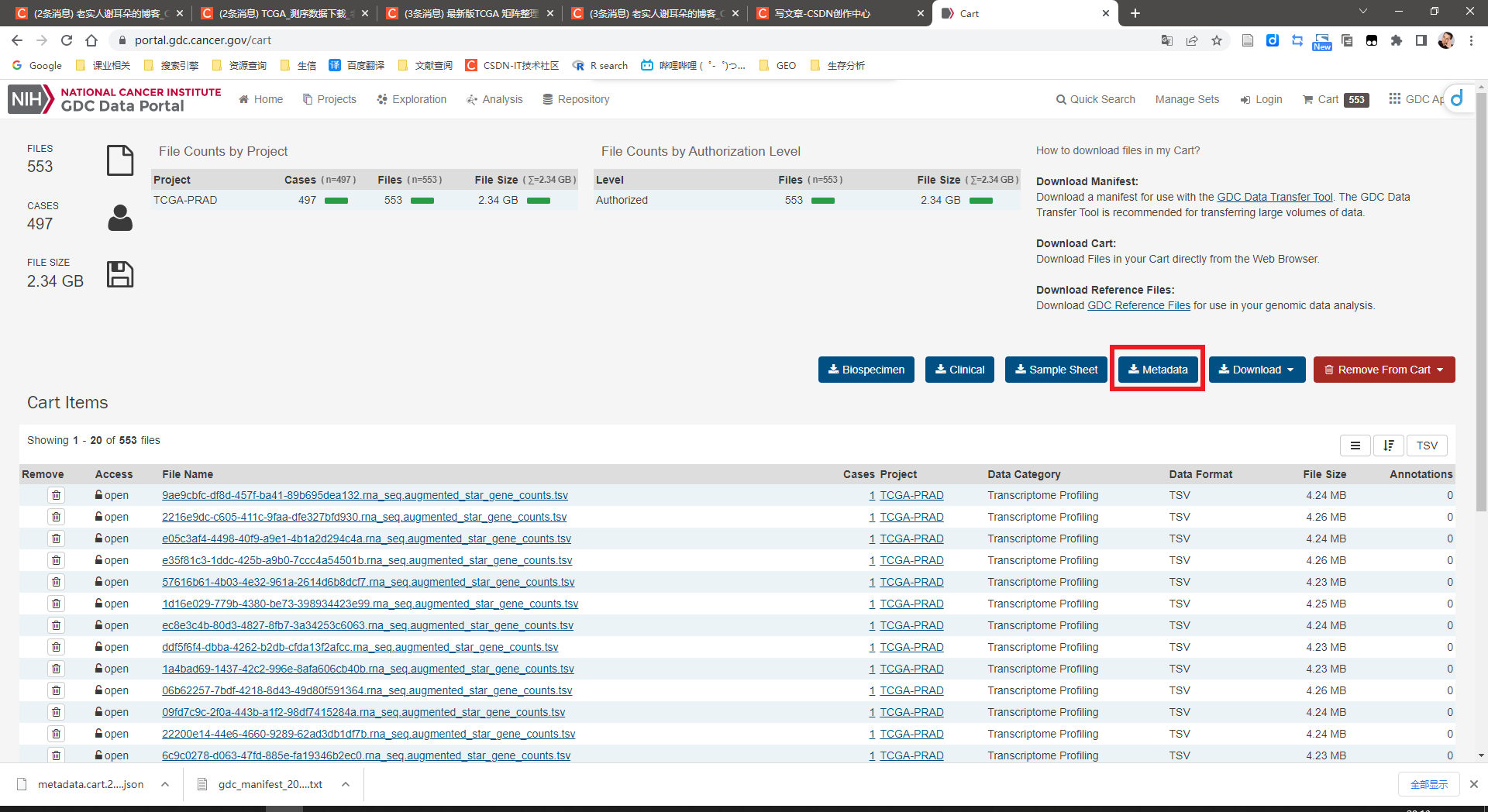
点击Metadata下载json文件
二、整合tsv文件、json文件到一个文件夹
1 右上角搜索“.tsv”
2 复制到新文件夹里面,整理如下:
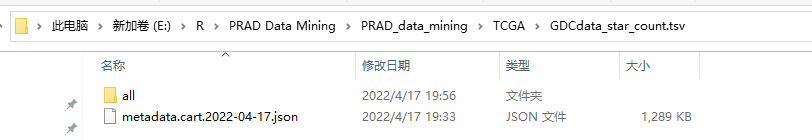
本机路径:'E:/R/PRAD Data Mining/PRAD_data_mining/TCGA/GDCdata_star_count.tsv/all/'
三、提取count矩阵
rm(list=ls())
options(stringsAsFactors = F)
library("rjson")1 整理“all”中的文件
result <- fromJSON(file = "E:/R/PRAD Data Mining/PRAD_data_mining/TCGA/GDCdata_star_count.tsv/metadata.cart.2022-04-17.json")
metadata <- data.frame(t(sapply(result,function(x){
id <- x$associated_entities[[1]]$entity_submitter_id
file_name <- x$file_name
all <- cbind(id,file_name)
})))
rownames(metadata) <- metadata[,2]
t_dir <- 'E:/R/PRAD Data Mining/PRAD_data_mining/TCGA/GDCdata_star_count.tsv/all/'
t_samples=list.files(t_dir)
sampledir <- paste0(t_dir,t_samples) #各个文件路径View(metadata)
metadata中记录barcode文件名的对应关系
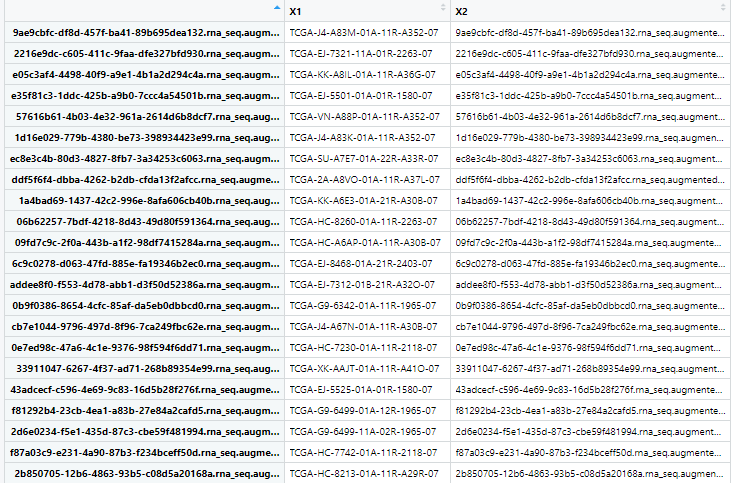
example <- data.table::fread('E:/R/PRAD Data Mining/PRAD_data_mining/TCGA/GDCdata_star_count.tsv/all/005d2b9e-722c-40bd-aa5c-bd4e8842cb04.rna_seq.augmented_star_gene_counts.tsv',data.table = F)#读入一个tsv文件,查看需要的列数,“unstranded”
raw <- do.call(cbind,lapply(sampledir, function(x){
rt <- data.table::fread(x,data.table = F) #data.table::fread函数
rownames(rt) <- rt[,1]
rt <- rt[,4]###第4列为“unstranded”
}))View(example)
载入一个例子,需要的 ”unstranded count” 值数据是第4列;同时第5行之后才可读
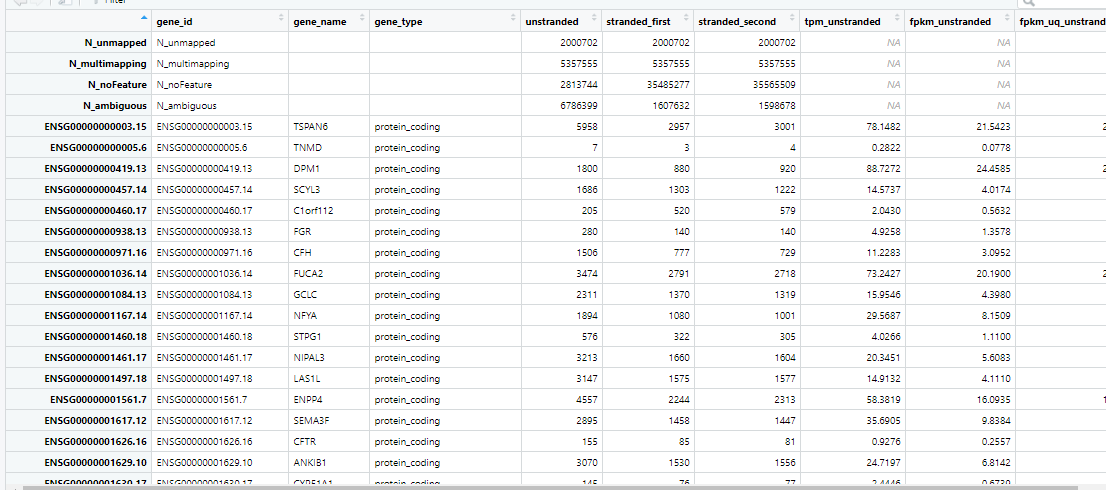
View(raw)
行名、列名未注释

2 替换列名和行名
重点说一下sapply(strsplit(sampledir,'/'),'[',8) #数字可选。意思是将每个sampledir按“/”分隔后,取第“8”个,对应的是tsv文件的文件名。比方说我的sampledir[1]:
'E:/R/PRAD Data Mining/PRAD_data_mining/TCGA/GDCdata_star_count.tsv/all/005d2b9e-722c-40bd-aa5c-bd4e8842cb04.rna_seq.augmented_star_gene_counts.tsv'
按“/”分隔后,第“8”个部分是tsv文件的文件名,所以我这里的参数为“8”
colnames(raw)=sapply(strsplit(sampledir,'/'),'[',8) #数字可选,'8'为文件名005d2b9e-722c-40bd-aa5c-bd4e8842cb04.rna_seq.augmented_star_gene_counts.tsv
rownames(raw) <- example$gene_id ##行名
raw_t <- t(raw)
t_same <- intersect(rownames(metadata),rownames(raw_t))
dataPrep2 <- cbind(metadata[t_same,],raw_t[t_same,])
rownames(dataPrep2) <- dataPrep2[,1]
dataPrep2 <- t(dataPrep2)
dataPrep2 <-dataPrep2[-c(1:6),] #dataPrep2为未注释count矩阵View(raw)
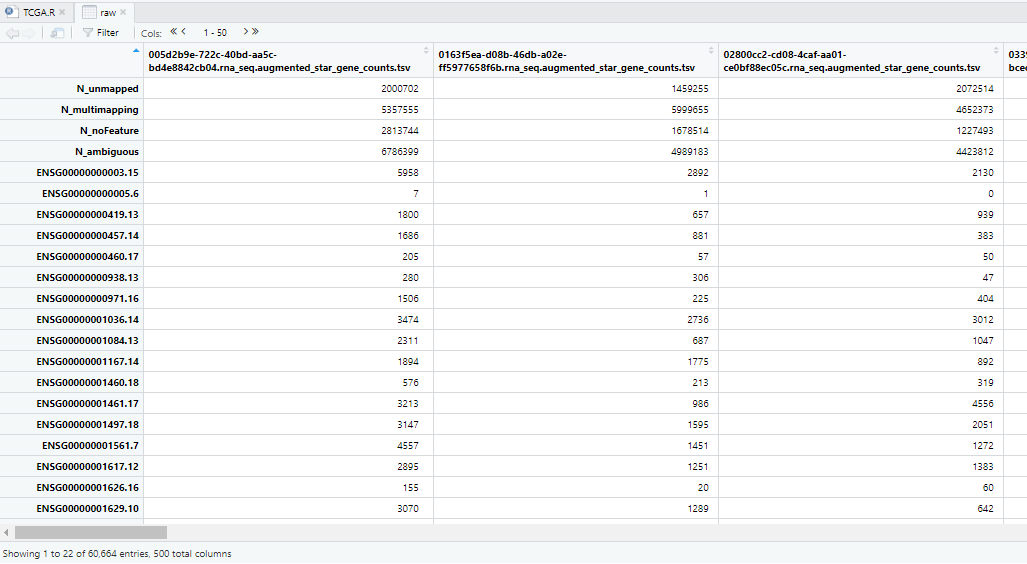
View(dataPrep2)
dataPrep2为未基因注释的count矩阵,格式为“matrix”
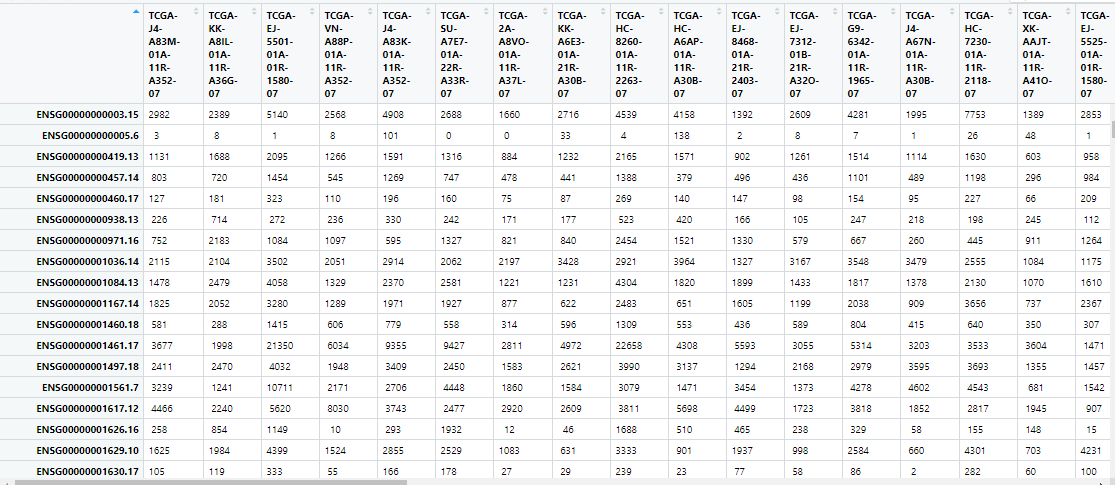
3 dataPrep2中数据类型为“character”,需要转为“numeric”
puried_data=apply(dataPrep2,2,as.numeric)View(puried_data)
行名没了
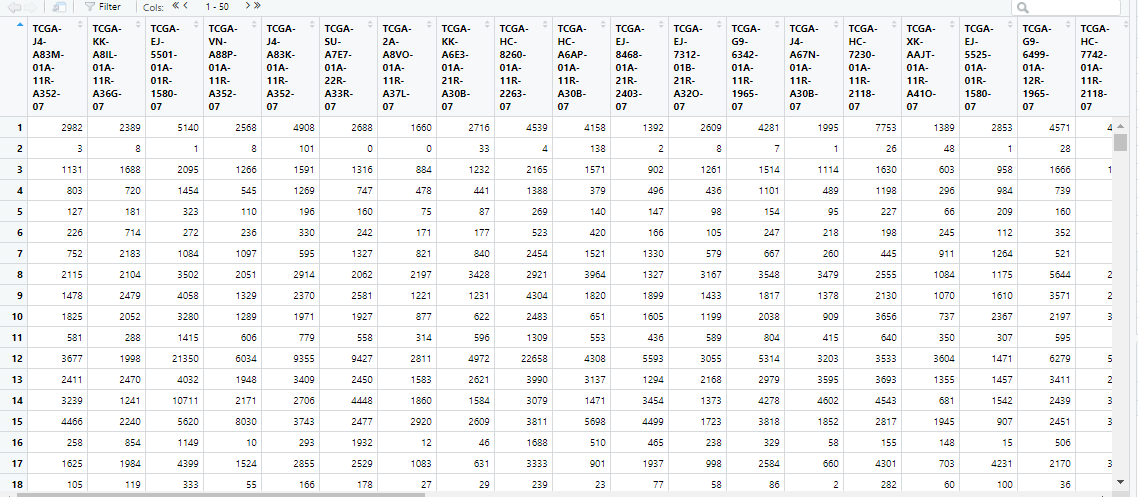
4 基因注释
puried_data的格式为“matrix”:“matrix”行名可以重复,因此直接替换行名。
此处仍然借用example进行基因注释
rownames(puried_data)=example[5:nrow(example),'gene_name']
View(puried_data)
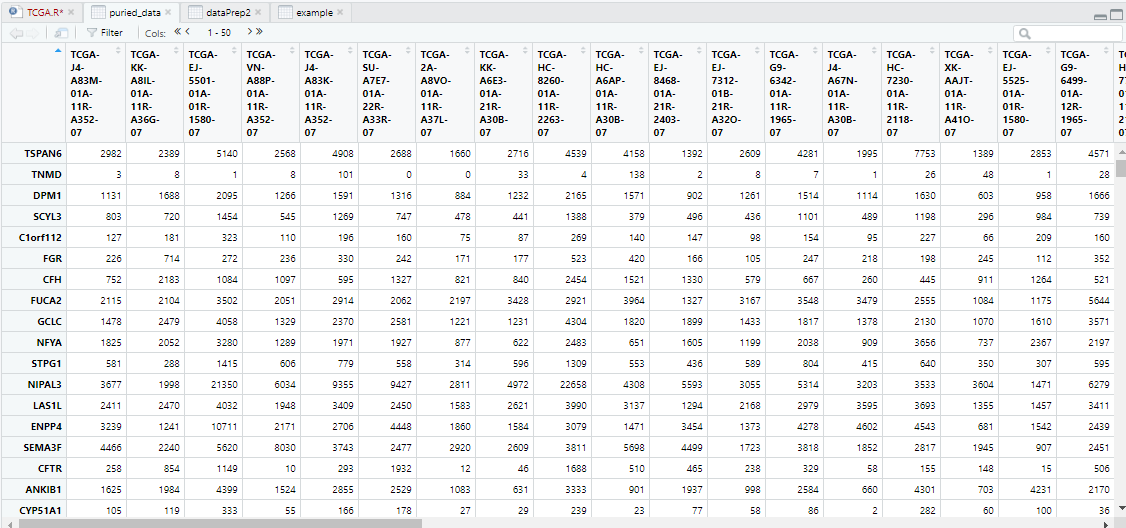
dim(puried_data)
[1] 60660 490
5 去除重复基因名
取行平均count值最大的行
t_index=order(rowMeans(puried_data),decreasing = T)#计算所有行平均值,按降序排列
t_data_order=puried_data[t_index,]#调整表达谱的基因顺序
keep=!duplicated(rownames(t_data_order))#对于有重复的基因,保留第一次出现的那个,即行平均值大的那个
puried_data=t_data_order[keep,]#得到最后处理之后的表达谱矩阵
dim(puried_data)
[1] 59427 490
6 预处理
需要注意DESeq2包要求数据未经过标准化
dataFilt =puried_data[rowMeans(puried_data)>10,] #剔除低表达基因
write.csv(dataFilt,file = "dataFilt.csv",quote = FALSE)
得到 dataFilt 后就可以正常分析了!
方法2(未成功)
尝试使用TCGAbiolinksR包
library("BiocManager")
library("TCGAbiolinks")
library("SummarizedExperiment")
BiocManager::install("BioinformaticsFMRP/TCGAbiolinksGUI.data")
BiocManager::install("BioinformaticsFMRP/TCGAbiolinks")
#比BiocManager::install(TCGAbiolinks)安装更新版的TCGAbiolinks包query <- GDCquery(project = "TCGA-PRAD", #项目名
data.category = "Transcriptome Profiling",
data.type = "Gene Expression Quantification",
workflow.type = "STAR - Counts")
t_samplesDown <- getResults(query,cols=c("cases"))
#从sampleDown检索出TP
t_dataSmTP <- TCGAquery_SampleTypes(barcode = t_samplesDown,
typesample = "TP") #可选
#筛选完成的query
queryDown <- GDCquery(project = "TCGA-PRAD",
data.category = "Transcriptome Profiling",
data.type = "Gene Expression Quantification",
workflow.type = "STAR - Counts",
barcode = t_dataSmTP)
#下载GDCquery的结果
GDCdownload(queryDown,
method = "api",
directory = "GDCdata_star_count",
files.per.chunk = 6) #网速慢可以设小一点
dataPrep1 <- GDCprepare(query = queryDown,
save = T,
directory = 'GDCdata_star_count', #默认为“GDCdata”
save.filename ="dataPrep1_PRAD_star.rda")
dataPrep2 <- TCGAanalyze_Preprocessing(object = dataPrep1,
cor.cut = 0.6
#datatype = "HTSeq - Counts") View(dataPrep2)
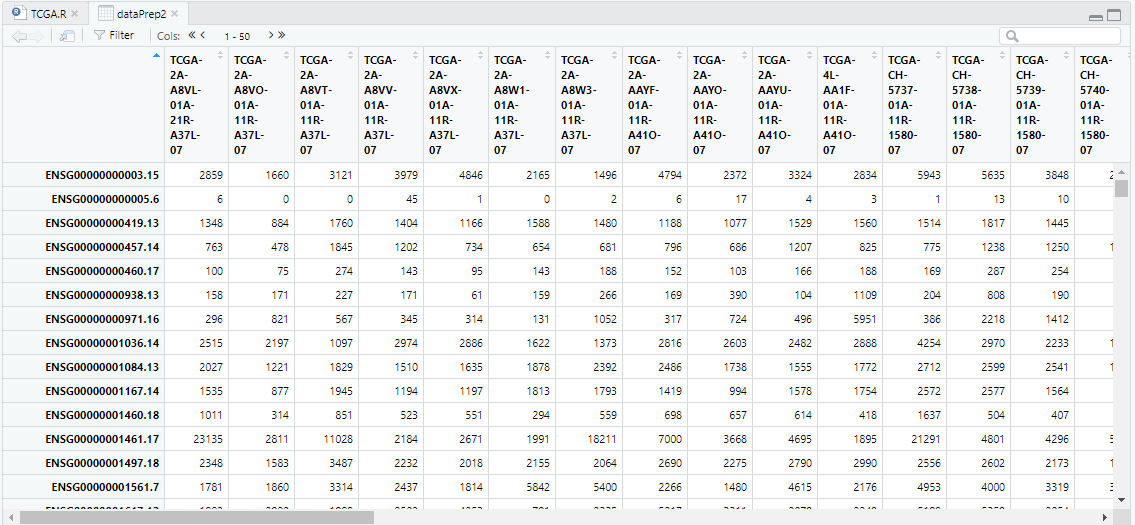
t_purityDATA <- TCGAtumor_purity(colnames(dataPrep1), 0, 0, 0, 0, 0.6)
t_purity.barcodes<-t_purityDATA$pure_barcodes #肿瘤样本barcodes,为“character”
t_normal.barcodes<-t_purityDATA$filtered #正常组织的数据barcodes,为“character”
puried_data <-dataPrep2[,t_purity.barcodes]#筛选后数据
rownames(puried_data)<-rowData(dataPrep1)$external_gene_name rownames(puried_data)<-rowData(dataPrep1)$external_gene_name (基因注释步骤)
View(puried_data)
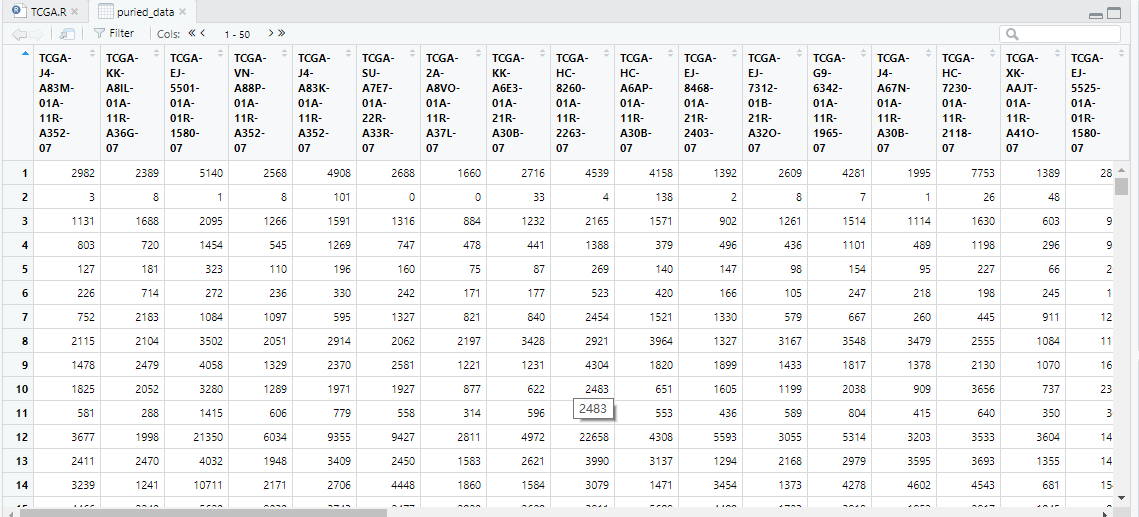
基因注释未成功,暂时没有找到解决方法



















 3485
3485

 被折叠的 条评论
为什么被折叠?
被折叠的 条评论
为什么被折叠?
 到【灌水乐园】发言
到【灌水乐园】发言











