enricher()函数
描述
用法
enricher(gene, pvalueCutoff = 0.05, pAdjustMethod = "BH", universe, minGSSize = 10, maxGSSize = 500, qvalueCutoff = 0.2, TERM2GENE, TERM2NAME = NA)
参数
- gene:一个“geneID”向量,使用哪种ID类型可以自定义,但需要和TERM2GENE保持一致
- pAdjustMethod:一般选择 "BH" 或 "fdr",BH较严格,fdr较温和(计算的q小些)
- pvalueCutoff:p阈值
- qvalueCutoff:q阈值
- TERM2GENE:一个两列的数据框,一列为“pathID”,一列为“geneID”
- TERM2NAME:一个两列的数据框,一列为“pathID”,一列为“pathName”
- 本质上只要有 pathName、geneID 二者间的对应关系就可以对任意自定义通路数据集进行富集分析,如对 Human Phenotype Ontology 进行通路富集等
代码
gene_entrezID
[1] 109 10398 1026 100289462 100289462 100289462 100289462
[8] 1026 111 108 10000 10398 10459 10152
[15] 100137049 11025 11025 11025 11025 10912
gene_symbol
[1] "ADCY3" "MYL9" "CDKN1A" "DEFB4A" "DEFB4B" "DEFB4A" "DEFB4B" "CDKN1A" "ADCY5" "ADCY2"
[11] "AKT3" "MYL9" "MAD2L2" "ABI2" "PLA2G4B" "LILRB3" "LOC102725035" "LOC107987425" "LOC107987462" "GADD45G"
pathset <-read.csv(file = "keggPath_entrezID_ensembleID.csv", header=T, check.names=FALSE)
term2gene = data.frame(Term = pathset$kegg_ID, Gene = pathset$entrez_ID)
term2name = data.frame(Term = pathset$kegg_ID, Name = pathset$pathway_way)
ENRICH = enricher(gene = gene_entrezID,pvalueCutoff = 0.9,pAdjustMethod = 'fdr',qvalueCutoff = 0.9,TERM2GENE = term2gene,TERM2NAME = term2name)
ENRICH.df = as.data.frame(ENRICH)
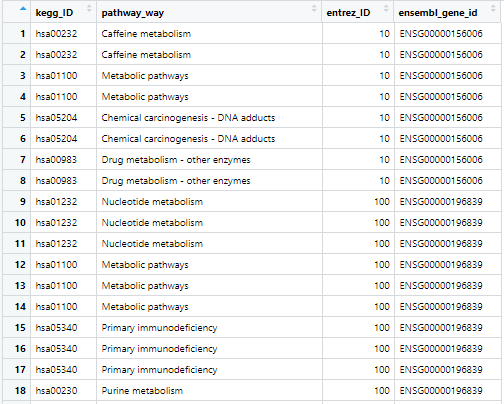 keggPath_entrezID_ensembleID.csv
keggPath_entrezID_ensembleID.csv
- 读取的 keggPath_entrezID_ensembleID 包含了 pathID、pathName 和 geneID 的对应关系
 ENRICH.df
ENRICH.df
- 得到的富集分析结果和直接使用 enrichGO、enrichKEGG 函数的分析结果相似



























 617
617

 被折叠的 条评论
为什么被折叠?
被折叠的 条评论
为什么被折叠?
 到【灌水乐园】发言
到【灌水乐园】发言











