







 文章介绍了SPIRAL模型,一种用于整合和对齐不同实验、条件和技术的空间转录组数据的深度学习方法。对比了其他算法的局限性,并详细阐述了GraphSAGE编码器、解码器以及生物鉴别和噪声分类网络。SPIRAL通过改进解决了批次效应和数据对齐问题,未来有多种可能的扩展方向。
文章介绍了SPIRAL模型,一种用于整合和对齐不同实验、条件和技术的空间转录组数据的深度学习方法。对比了其他算法的局限性,并详细阐述了GraphSAGE编码器、解码器以及生物鉴别和噪声分类网络。SPIRAL通过改进解决了批次效应和数据对齐问题,未来有多种可能的扩展方向。
目录
3.3 生物鉴别网络(biology discriminator)
0 参考文献
1 算法比较
对于空间转录组数据整合或者数据对齐的算法,我已经在前面的博客介绍过几个,这里就不再赘述空间转录组学的背景了。
对于SPIRAL所完成的任务——空转数据整合和对齐,已经并不是一个新的任务了,但是SPIRAL在整体的表现上相比部分其他算法有所突破。
在论文中大致介绍了一些其他相关算法存在的局限性,然而有些方法我并不了解,因此在这里只依据论文中的给出的部分算法局限性进行介绍。
1)BASS:BASS算法旨在使用贝叶斯分层方法进行多sample的空间转录组数据整合,从而同时实现统一聚类和区域识别。但是首先,BASS并没有对于表达矩阵的低维表示(没有消除了批次效应的表示);其次,在对来自不同技术的空转数据进行统一聚类时性能并非最优。
2)GraphST:GraphST算法对已通过其它算法实现切片对齐的数据进行批次校正。局限在于该效果很大程度上依赖于对齐的准确性。
3)DeepST:DeepST算法利用图神经网络进行表征学习,然后使用域对抗神经网络对这些表征进行批处理效果去除。其局限性在于无法获得不受批次效应影响的基因表达数据,并且其训练方法阻碍了不同sample之间的整合。
4)STAligner:STAligner通过整合STAGATE算法和MNN算法实现数据整合和切片对齐,这种方法的对齐很大程度上依赖MNN对,因此可能会忽略来自同一空间域的非MNN对,并错误地包括属于不同空间域的MNN对。
5)PASTE:PASTE算法同时使用基因表达矩阵和空间坐标矩阵实现数据对齐和整合。然而对于覆盖程度较低的两个切片,PASTE的结果将不尽如人意,这意味着PASTE无法在多sample之间进行数据整合。
SPIRAL则克服了上述局限性。
2 SPIRAL模型结构
SPIRAL由SPIRAL-integration和SPIRAL-alignment两部分组成。
SPIRAL-integration通过结合GraphSAGE网络和域适应网络(domain adaptation network),使用基因表达数据和空间位置关系数据作为输入,来纠正批次效应。
SPIRAL-integration部分由四个神经网络构成,分别为1)作为编码器的GraphSAGE网络,2)噪声分类网络,3)生物鉴别网络,这两者用于将低维表示分解为噪声部分和生物部分,4)用于从低维表示重建基因表达的解码器网络。如下图所示。
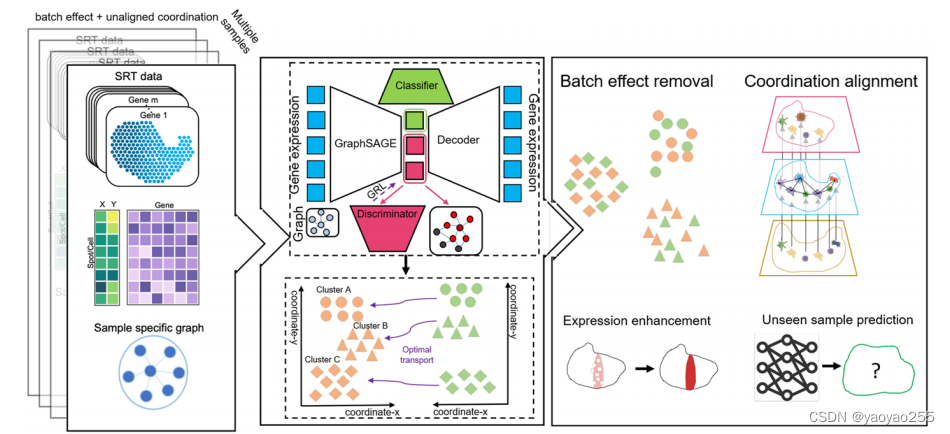
SPIRAL-alignment通过SPIRAL-integration学习的表示的聚类结果,将一个样本的共享类内的点分分配到参考样本的相应位置。
3 具体算法
3.1 GraphSAGE(编码器)
GraphSAGE是一个基于深度神经网络的图节点嵌入框架。他通过节点特征(基因表达矩阵中的一行)和图结构(根据相对位置关系构建的图)得到对于节点的低维表示。
首先,对于每个切片数据构建无向图。假设某切片点数为n,则该无向图可以表示为一个n*n的矩阵,当两个点之间存在边连接时,该位置设置为1,否则为0。判断两点之间是否应有边相连可以使用knn方法,将近邻点用边相连。
该编码器以基因表达矩阵和无向图作为输入,一共包含K层,其中第k层的节点表示通过下面公式计算。先将与该节点有边连接的节点的上一层表示通过某种方式集成(可以是取平均、池化、LSTM aggregator方式),再将得到的向量与上一层该节点的表示连接起来,乘上一个可学习矩阵,最后进行ReLU非线性激活。
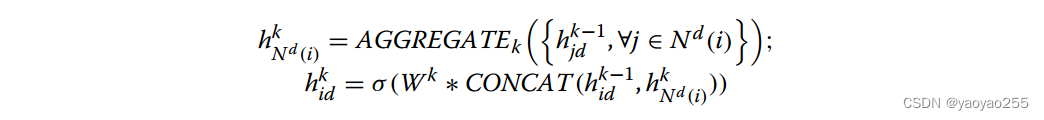
最后得到低维表示如下
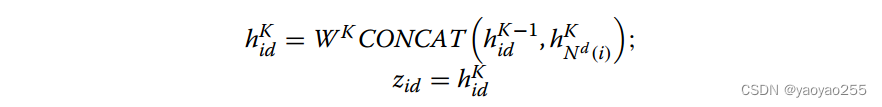
损失函数如下。描述了学习到的低维表示重建原图的能力,具体通过增大邻近节点表示相似程度(第一部分)并减小非邻近节点表示相似程度(第二部分)实现。其中ρ是Sigmoid函数。
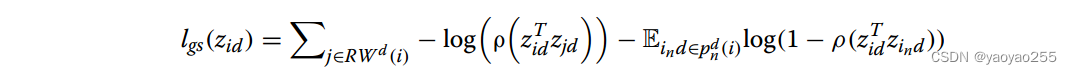
3.2 解码器
解码器以节点的低维表示作为输入并重建原基因表达数据(节点特征)。第k-1层输出由第k层输出计算得到,计算方式如下
![]()
最终重建的基因表达数据如下
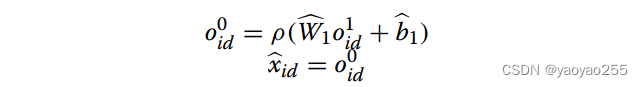
解码器的损失函数如下,这种累加作用于M个基因。
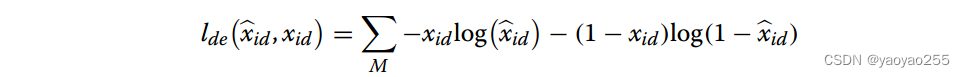
3.3 生物鉴别网络(biology discriminator)
这个鉴别网络从编码器输出的低维表示中选取更低维的一部分作为输入,以批次ID作为输出,记为y_id。生物鉴别网络目的是通过训练生成器(GraphSAGE)通过采用梯度反转层(GRL)欺骗训练良好的鉴别器,使批次ID与低维表示抽取的部分无法区分。
生物鉴别器的输出通过下面方式获得
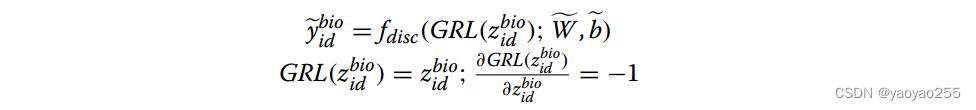
其中f_disc是一个多层非线性函数,GRL是梯度反转层,前向传播时输入输出相同,反向传播时输入输出相反。
这部分的损失函数如下,其中θ是交叉熵函数。
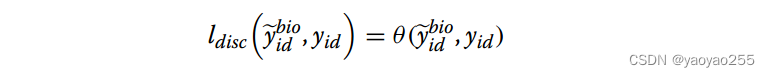
3.4 噪声分类网络(noise classifier)
噪声分类器使用低维表示去掉生物鉴别器选取部分的剩余部分作为输入,以批次ID作为输出。这部分的目的是使可以通过选取出的这一部分,很好地区分不同批次,这样可以通过设置低维表示中的这一部分为0来消除批次效应。
输出与损失函数如下
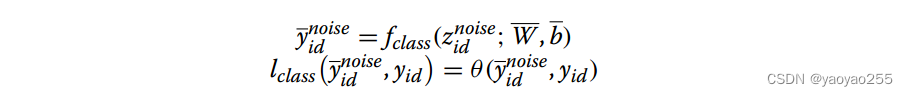
3.5 总体损失函数
SPIRAL-integration部分的总体损失函数是四个独立损失函数的加权,其中M为基因数量,λ和γ为超参数。
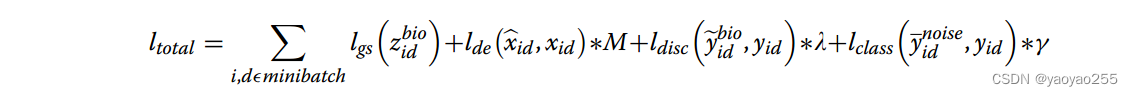
3.6 切片坐标对齐
要实现切片坐标对齐,首先需要选取一个切片作为参考切片,将其他切片对齐到该切片上。
使用与PASTE类似的方式计算Gromov-Wasserstein距离,不同点在于,SPIRAL-alignment的该距离只在聚类后的同类别之间计算,而PASTE则没有聚类这一步,直接使用整个切片进行计算。
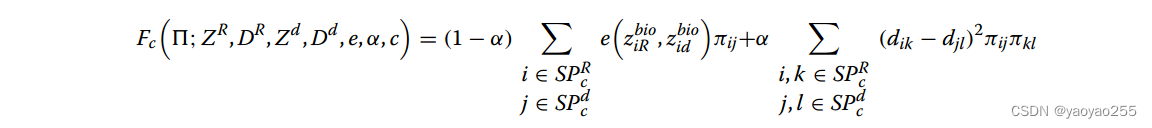
其中c是第c个聚类,R代表参考切片,d为要进行对齐的切片号。上式的具体含义可以参考PASTE博客。
这之后,可以通过如下方式计算节点在参考切片坐标系中的新坐标。第d个切片第c个聚类中的点j新坐标如下
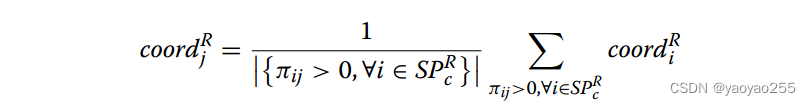
最后,还需要进行主要包括旋转和平移坐标变换,以属于切片间共有的聚类的点的位置作为landmark,来确定切片特有的聚类的点的位置。
4 改进方向
文章结尾作者提出了SPIRAL的几点改进方向:
1)SPIRAL可以进一步结合组织图像、空间位置和基因表达数据,从而同时消除基因表达和图像的批次效应。
2)SPIRAL-alignment可以被改进以使得不同的cluster之间具有较为均匀的对齐。
3)SPIRAL可以被推广到更高分辨率的数据,以实现更精确的结构描绘。
4)可以通过并行计算或分布式学习来加速训练过程。
5)SPIRAL可以扩展其整合多组学空间数据的能力。

















 2280
2280

 被折叠的 条评论
为什么被折叠?
被折叠的 条评论
为什么被折叠?
 到【灌水乐园】发言
到【灌水乐园】发言









