






 本文介绍了10XVisium技术,一种用于研究组织基因表达的空间转录组学方法。详细解读了芯片结构,包括探针设计和功能,以及操作流程,包括样本准备、组织透化、cDNA合成和数据处理。对生物信息学初学者理解该技术有很大帮助。
本文介绍了10XVisium技术,一种用于研究组织基因表达的空间转录组学方法。详细解读了芯片结构,包括探针设计和功能,以及操作流程,包括样本准备、组织透化、cDNA合成和数据处理。对生物信息学初学者理解该技术有很大帮助。
目录
10X Visium是一种空间转录组学技术,用于研究组织和细胞在其原位空间中的基因表达模式。这种技术允许研究者同时获得组织中数千个基因的表达信息,并将其与组织的空间结构相对应。
AI for Science飞速发展的当下,机器学习在生物信息领域的应用备受关注,空间转录组学是其中很重要的一部分,对于没有生物信息学背景的人,上手使用数据往往是一个难点。理解相关技术的原理可能能够帮助大家理解数据。
1 芯片结构
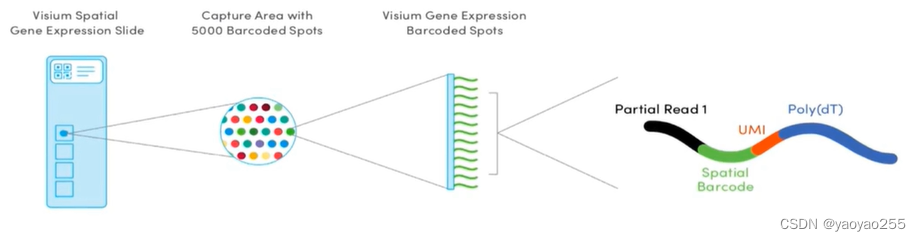
上图为10x Visium技术使用到的芯片(slide)结构如上图所示,用于捕获细胞切片中的mRNA。
每个芯片(左1)包含四个捕获区,即芯片上的四个正方形区域,每个捕获区大小为8mm*8mm,其中真正有用的部分大小为6.5mm*6.5mm。
每个正方形区域有约5000个spot组成(左2),单个spot的直径为55μm,相邻spot圆心距为100μm。因此每个spot可以捕获多个细胞。对于捕获的细胞数,不同文章有不同的说法,这也与细胞大小、细胞间距等有关,但大致范围为几个到几十个之间。
从侧面看单个spot如图(右2)所示,每个spot上有数百万个探针。单个探针由四部分组成(右1)。
黑色区域Partial Read 1测序时所需要的部分引物序列。
绿色区域Spatial Barcode是用于确定探针位置的条码,相同每个探针都有一个该序列,同一个spot内所有探针的该序列相同,而不同spot之间该序列互不相同,以此可以确定表达产物的空间位置。
红色区域UMI是unique molecular identifier的缩写,每个spot中不同探针具有不同UMI,用于确定捕获到的转录本数量。
蓝色区域Poly(dT)有许多T碱基组成,与mRNA3'端的一连串A碱基序列互补,用于捕获mRNA。
2 操作流程
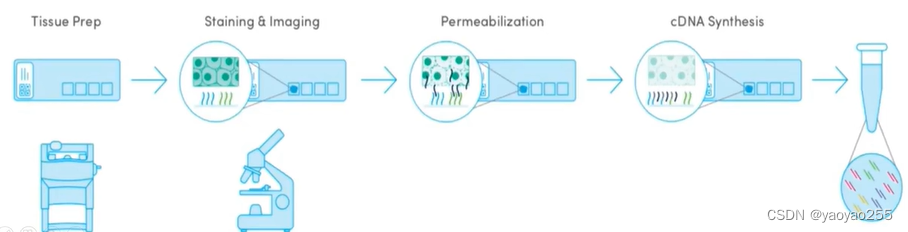
上图展示了10x Visium技术流程。
1)样本准备,将组织切片固定到捕获区(芯片正方形区域),进行HE染色及拍照
2)进行组织透化(permeabilization),破坏细胞,释放mRNA,是其与探针结合
3)由于mRNA不稳定,故将其逆转录成cDNA
4)将cDNA(探针和cDNA的结合物)洗脱,随后可以进行文库构建、测序及数据可视化。由于每个探针具有Spatial Barcode,因此即使不同spot的探针混在一起,也可以保留各自的空间位置信息
下面是另一个版本的原理图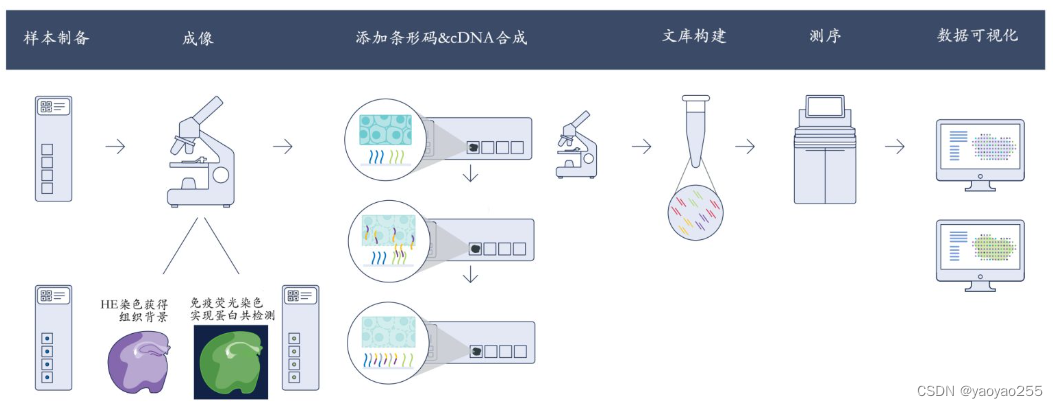

















 1万+
1万+

 被折叠的 条评论
为什么被折叠?
被折叠的 条评论
为什么被折叠?
 到【灌水乐园】发言
到【灌水乐园】发言









