






在完成差异基因表达分析之后,自然而然可以想到GSEA分析(gene set enrichment analysis,基因集富集分析),这个分析需要使用基因差异表达的信息和一系列基因集进行。分析结果是这个基因集的标准化富集分数和相应的p值。
注意,本文使用的是fgesa这个R包,不是常见的clusterprofiler这个包(因为博主clusterprofiler包里的GSEA分析不知道为啥有点寄)。
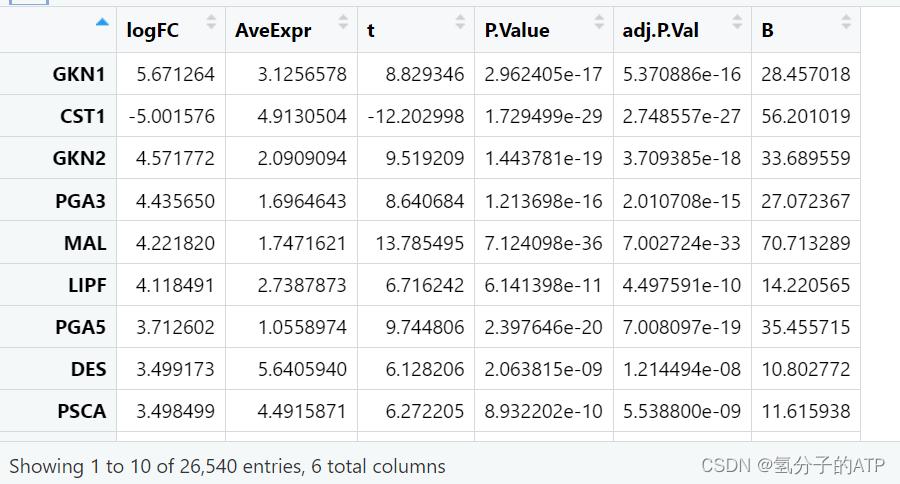
首先,先获取差异表达分析的结果,如果不清楚差异表达分析,可以先去前一篇文章看看要怎么操作(RNA-seq分析入门01——差异基因表达分析-CSDN博客),这里我们就直接使用上篇文章里通过limma获得的diff数据框了。diff数据框的结构如下所示。
首先,先加载以下R包:
library(clusterProfiler) #改基因SYMBOL为ENTREZID
library(fgsea) #GSEA分析
library(org.Hs.eg.db) #人类的基因SYMBOL和ENTREZID的对照数据
library(msigdbr) #保存了各种基因集的一个包
library(ggplot2) #绘图然后,我们要把基因SYMBOL转换为fgsea能够识别的ENTREZID,并且只保留基因的log2FC单列,并使用ENTREZID对其命名。
#读取差异表达数据(只要基因和logFC), diff移步limma结果
data<-cbind(rownames(diff),diff) #把行名上的基因SYMBOL加到diff的第一列
colnames(data)[1]<-'SYMBOL'
gene<-bitr(data[,1], fromType = 'SYMBOL',toType = 'ENTREZID', OrgDb = 'org.Hs.eg.db') #获得基因的SYMBOL和ENTREZID之间的对照表
data<-subset(data,rownames(data)%in%gene$SYMBOL) #删除转换名字后没有对应ENTREZID的基因
gene_data<-merge(data,gene, by.y='SYMBOL') #合并基因名字和diff
#整理gene_list, 格式为logFC, 以entrezid命名(只要一列logFC的数据,要用ENTREZID命名)
diff_list<-gene_data[,2]
names(diff_list)<-gene_data[,8]
#排序
diff_list<-diff_list[order(diff_list, decreasing = T)] #要按照logFC排序,不然之后会报错这个是diff_list的样式。按照log2FC排序的由ENTREZID命名的一列数据。

接下来是获取基因集列表,使用msigdbr数据库获取基因集。这里示例使用hallmark基因集。
glist <- msigdbr(species = 'Homo sapiens', category = 'H')#选择hallmark
gene_set<-split(x = glist$entrez_gene, f = glist$gs_name)#按entrezid分出基因集列表用到的基因集列表gene_set如下所示。
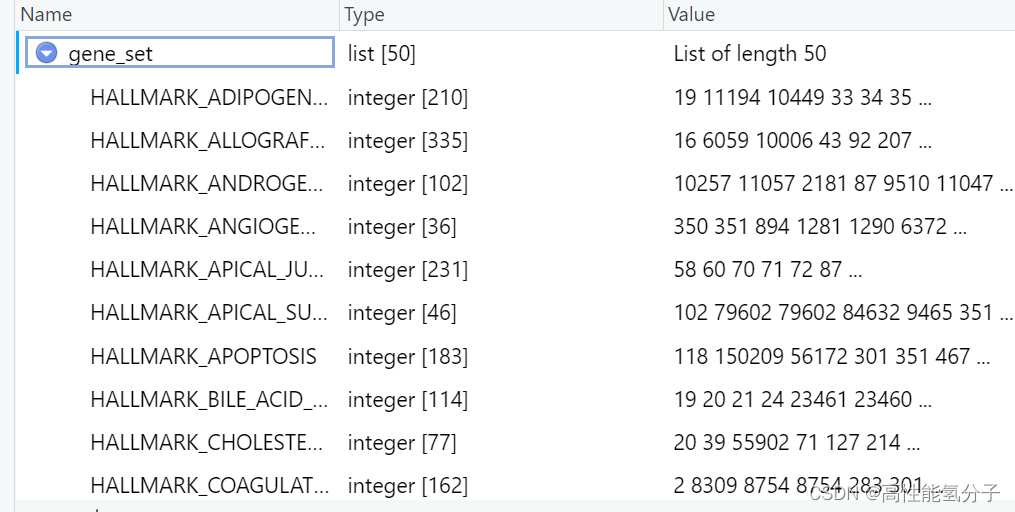
这里fgsea也可以使用自定义的基因集,但是要以如上所示的格式来构造列表,并且用你的通路的名字命名。
example<-list('your pathway'=c(1,2,3,4,5))#记得给通路命名,不然后面的都做不了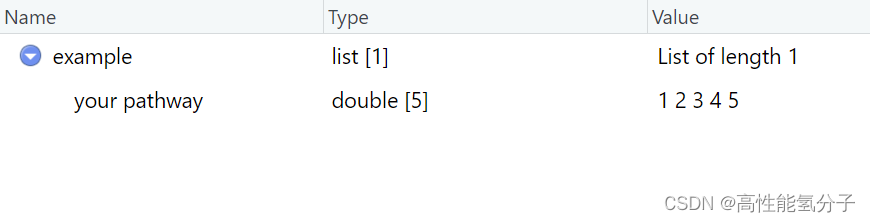
下一步就是运行fgsea了。
##运行GSEA分析
#diff_list最好使用全部基因的,反正一定要比gene_set的基因数量多
gsea_res <- fgsea(pathway=gene_set,stats=diff_list)gsea_res就是GSEA分析的结果,具体结果如下所示。
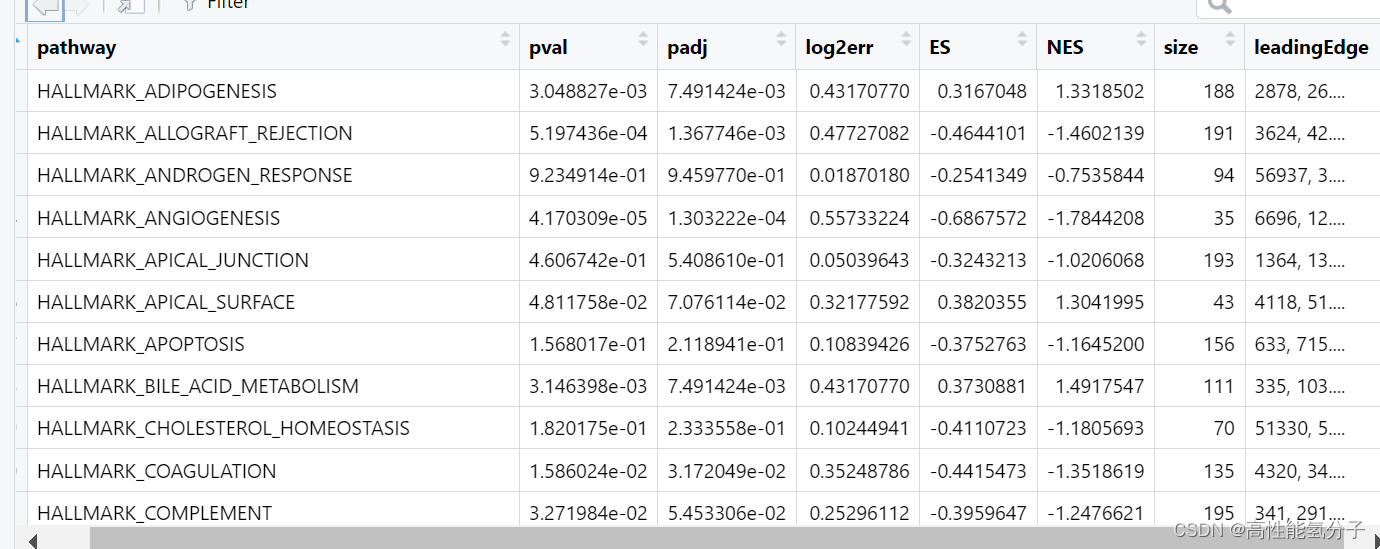
padj是假阳性率,越低越好,通常认为小于0.25即符合要求,pval是p值,通常认为小于0.05符合要求,NES是标准化富集分数,越高显示该通路富集越显著,通常NES>1或<-1为显著富集。
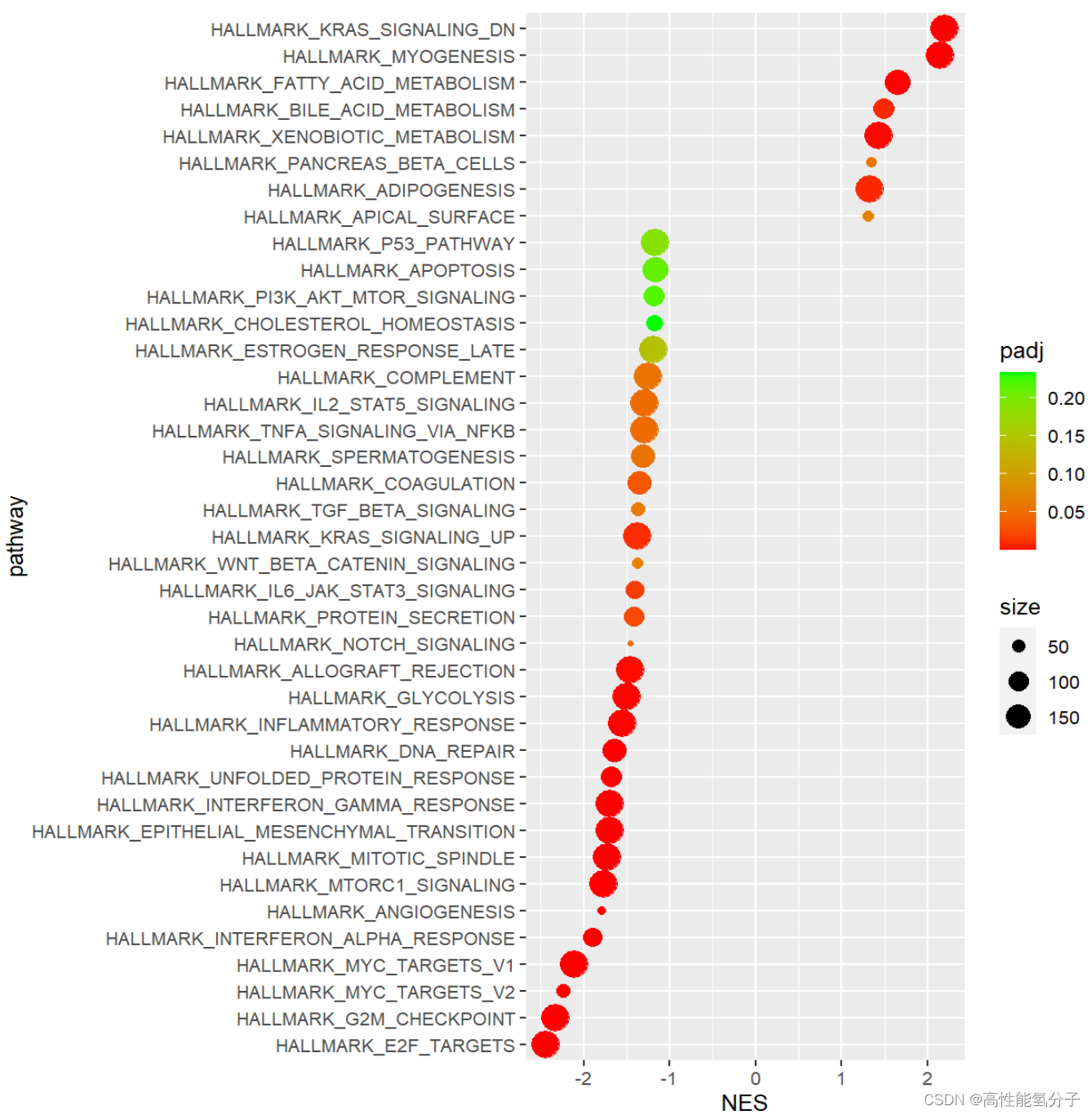
接下来就是作图,这里先演示对gsea_res的批量作图。
##批量作图
gsea_res<-subset(gsea_res, gsea_res$padj<0.25&abs(gsea_res$NES)>1)#常用pad<0.25,|NES|>1
gsea_res<-gsea_res[order(as.numeric(gsea_res$NES)),]#排序
y_name<-gsea_res$pathway #调整y轴刻度(不调整就不会按照NES的大小排序)
ggplot(gsea_res, aes(x=NES, y=pathway, color=padj, size=size))+geom_point()+
scale_y_discrete(limits=y_name)+scale_color_continuous(low='red',high='green')结果示意图如下所示
之后再看看对单个通路的GSEA结果怎么画。
这里示例使用的是HALLMARK_MYOGENESIS通路,先从gsea_res中选出这个通路的NES和padj,然后再标上。
##单个通路作图
#指定通路
pathway_name = 'HALLMARK_MYOGENESIS'
pval<-paste('p.adj = ',as.character(gsea_res[which(gsea_res$pathway==pathway_name), 3]))
nes<-paste('NES = ',as.character(gsea_res[which(gsea_res$pathway==pathway_name), 6]))
plotEnrichment(gene_set$HALLMARK_MYOGENESIS, diff_list)+
annotate('text',x=12000,y=0.45, label=pval)+annotate('text',x=12000, y=0.4, label=nes)#annotate的x和y调整文字的位置结果如下图所示
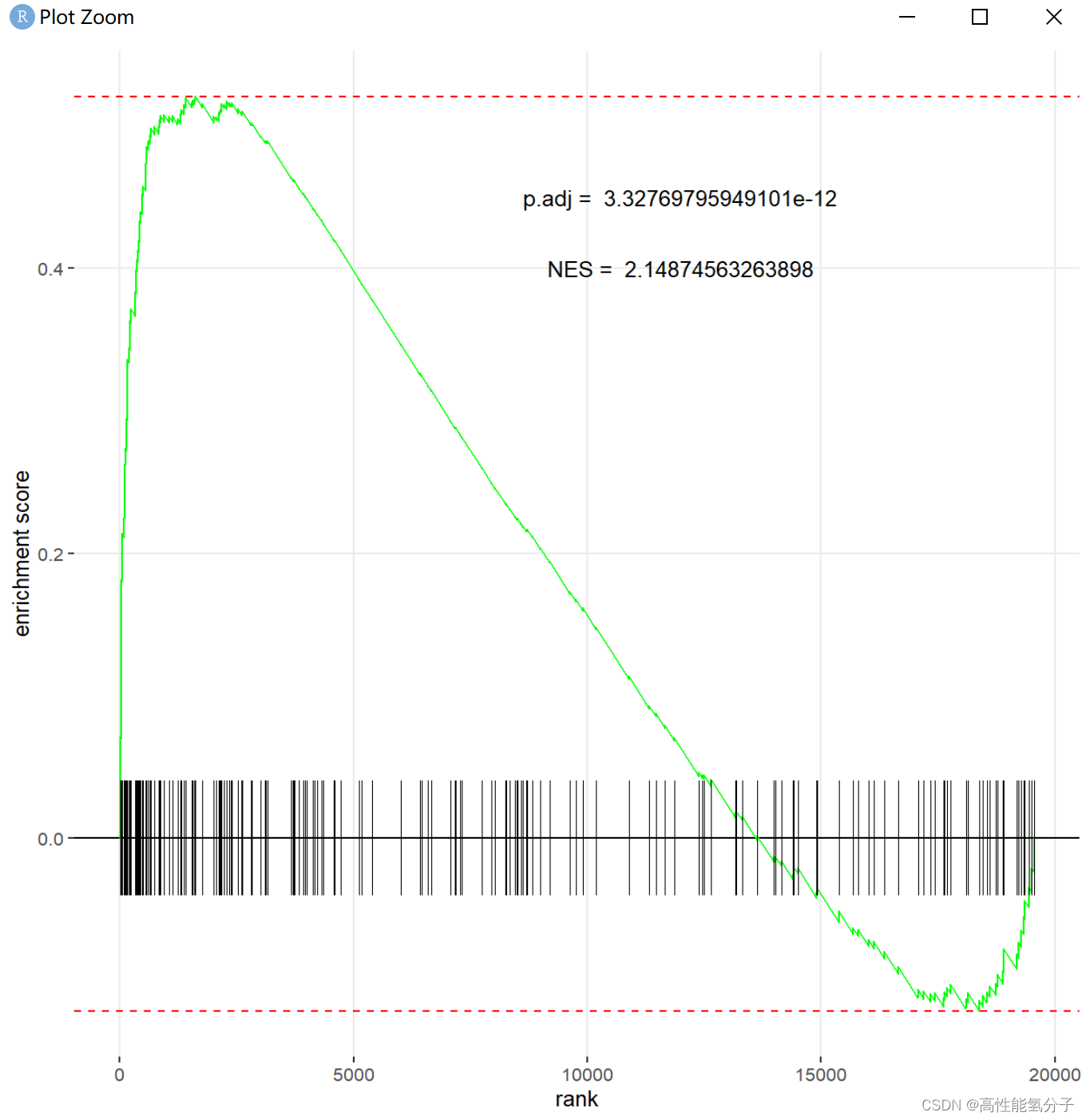
除了padj,nes等数值足够显著之外,GSEA的曲线要尽可能的平滑和呈现一个明显的趋势,增加差异表达的基因数量和基因集数量更有利于画出好看的GSEA图。

















 4560
4560

 被折叠的 条评论
为什么被折叠?
被折叠的 条评论
为什么被折叠?
 到【灌水乐园】发言
到【灌水乐园】发言









