






#编者按
本人为一名2025届毕业的农科生(写这篇文章的时候是大四)。从大二开始,在各种平台学习家族分析,因为是丐版Macbook 8G M1,真的踩过不少坑。专门重新写一个适合我们本科生阅读的家族分析教程,很多同学的毕业设计会选择家族分析,但是网上这么多教程,如何学习如何使用确实是个大问题。如果你还要考研,那可又是一个大冲击。不过放心,现在你们有这篇文章应该会很容易学习。祝愿大家都能顺利完成自己家族分析的毕业设计,取得优异的成绩。
本文章的特点在于,把 TBtools 和 linux 的办法放在一起比较,大家可以通过 TBtools 来理解 linux 指令。同时我也收集了很多与家族分析很相关的、必要的 linux 教程。 算是一篇小综述吧。
目录
【一】如何在 Macbook 上安装 linux 系统(可以先跳过)
【零】你想要如何完成家族分析?
这需要看看你是什么电脑。
如果你是拥有 16Gb 内存的win用户,那么市面上所有的软件和教程都非常适合你,你大可以不需要使用 linux 来完成你的分析。
但如果你和我一样是 Mac 丐版 8Gb 用户,那么很可能你有些TBtools功能是难以运行或者运行很慢。这个时候使用终端才行。关于终端的一些事情,我们可以慢慢说。
【一】如何在 Macbook 上安装 linux 系统(可以先跳过)
很幸运的是,macbook不需要安装linux系统就能够实现liunx的操作。至少在终端方面(Terminal),他们是差不多的。
这里引用知乎的一段话:“ MacOS 和 Linux 都可以在命令行中运行 Unix 命令。” MacOS 和 Linux 有什么区别?
首先我们需要打开终端,可以在搜索栏里搜索。或者进入“启动台”,找到“其他”,终端就在里面。这样子你就打开了终端,可以开始利用代码去执行你的命令。
#输入ls 可以展开当前目录。
(base) caimingzhe@bogon ~ % ls
Applications Virtual Machines.localized
Desktop anaconda3
Documents blender_file
Downloads config.log
E-Study config.status
Library documentation
Movies iCloud Drive (Archive)
libdivsufsort Music
Pictures profmark
Public src
PycharmProjects testsuite
#输入cd Desktop,你就可以在你的桌面进行生信分析了,非常方便你抓取文件和看文件里面的信息。
(base) caimingzhe@bogon ~ % cd Desktop
(base) caimingzhe@bogon Desktop %
#标题就变了,说明这是我们当前的目录。
#你可以手动创建一个文件夹,也可以使用指令。mkdir 就是 make directory
mkdir gene_analysis
cd gene_analysis
#你再把所需要分析的文件放进去,或者在这个文件夹里使用 wget 指令。
wget http://你基因组文件的下载链接
#但是这个下载可能不会太快,如果你是下载的国外网站上的基因组,比如ensembl或者NCBI,这个办法会很慢。
#这种情况我建议把链接粘贴到迅雷里下载,会快速很多。终端要安装conda(必须必须必须!)
conda真的太重要的,安装conda之后基本上全部的软件你都可以使用 conda install your_software_name 这个指令来安装。所以我才说是“必须”。
建议是安装miniconda,因为小一些,更加方便,Anaconda 本身要 1Gb,mini 则是 500Mb。
给你的 MacOS 装载 x64 虚拟环境
部分软件无arm64 构架,比如做共线性圈图的Circos和做多序列比对的ClustalW。
#查询电脑是x64架构还是arm64架构
uname -a
#conda构建x64虚拟环境,x64env是环境名称,你可以改成别的
CONDA_SUBDIR=osx-64 conda create -n x64env python=3.8
conda activate x64env
conda config --env --set subdir osx-64【二】基因家族分析都有哪些步骤?
如果你是一个学生,大概率你需要找导师要这个家族名称。当然,如果老师让你自己选择,你就在各种论文里找找看有哪些家族比较热门(明星家族)。
根据制作难度,在后面你可以看到星号,很明显进化树和共线性分析是最麻烦的部分。
- 感兴趣的家族基因名称 *
- 筛选出候选基因 *
- 构建多物种进化树(phylogenetic tree)*****
- 共线性分析(物种内和多物种)*****
- 分析基因结构 *
- motif预测 *
- 染色体定位 *
- 顺势作用元件(启动子)预测 **
- 亚细胞定位预测 *
- GO/KEGG分析(GO和KEGG是不一样的,一般都需要分析一下,但是在论文里面看到的不多)***
TBtools 可以完成上面所有的分析,但是!如果你是丐版 Macbook Air M1,你的 TBtools 很可能跑不出来共线性分析。这就很难办了,所以回归到最最最最最最基本的linux来分析就很重要了。
TBtools是一个很好的入门软件,功能十分强大。但是界面操作不可避免的缺点就是,很难批量操作,linux 在这方面有必然的优势。我会把TBtools和linux的操作放在一起比较。大家可以感受一下各自的优势。
【三】候选基因家族的筛选(BLAST + Hmm)
#前期文件准备
我们需要我们自己物种基因组的文件
- 染色体序列文件(your_species.dna.toplevel.fa)
- 基因注释文件gff3 (your_species.gff3)
- 蛋白质序列文件 (your_species.pep)
不知道这些文件是什么?可以看看这一篇
都2024年了,如何快速入门基因家族分析?|所需文件介绍
关于如何在TAIR和Ensembl Plant里下载这些文件可以参考一下文章。
一、TAIR 获取拟南芥中此家族基因序列
以我做的家族基因为例,半乳糖转移酶 (galactosyltransferase)。非常尴尬的是,TAIR 搜索不到任何这个家族基因的信息(暂时)。之后我查找文献发现这个酶的简称是GALT,搜索GALT就可以查到这些蛋白质。大概有14个蛋白质,一部分按照GALT命名好了,还有一部分具有半乳糖转移酶的功能但是有别的命名,别怕,这些也在我们的考虑范围内。
有的同学会问,如果在蛋白质文件里用 linux 的 grep 指令去搜索出更多的该怎么办?其实这个我也干过。
#比如这个pep文件的格式
>AT5G16970.1 pep chromosome:TAIR10:5:5575973:5578086:-1 gene:AT5G16970 transcript:AT5G16970.1 gene_biotype:protein_coding
transcript_biotype:protein_coding gene_symbol:AER description:alkenal reductase [Source:NCBI gene (formerly Entrezgene);Acc:831560]
MTATNKQVILKDYVSGFPTESDFDFTTTTVELRVPEGTNSVLVKNLYLSCDPYMRIRMGK
PDPSTAALAQAYTPGQPIQGYGVSRIIESGHPDYKKGDLLWGIVAWEEYSVITPMTHAHF
KIQHTDVPLSYYTGLLGMPGMTAYAGFYEVCSPKEGETVYVSAASGAVGQLVGQLAKMMG
CYVVGSAGSKEKVDLLKTKFGFDDAFNYKEESDLTAALKRCFPNGIDIYFENVGGKMLDA
VLVNMNMHGRIAVCGMISQYNLENQEGVHNLSNIIYKRIRIQGFVVSDFYDKYSKFLEFV
LPHIREGKITYVEDVADGLEKAPEALVGLFHGKNVGKQVVVVARE
#注意到description,会写上这个蛋白质可能的功能
#所以我当时写的指令是
grep '^>' ath.pep | grep 'GALT' | sed 's/>//' > GALT.txt
grep '^>' ath.pep | grep 'Galactosyltransferase' | sed 's/>//' > Gala.txt
grep '^>' ath.pep | grep 'galactosyltransferase' | sed 's/>//' > gala.txt
cat GALT.txt Gala.txt gala.txt > mixed_galt.txt
#解读一下这个指令
# ^> 指以>为开头,并非多此一举,有的氨基酸序列里会出现GALT。
# cat 可以把这三个文件合并成一个文件, 如果你文件夹里只有这三个文件,你也可以用
# cat *.txt > mixed_galt.txt
# 星号代表任意字符
# sed 's/>//' 可以去除行里面的 >号
这样子我就得到了全部的描述,但是这里面的 GALT 基因是有重合的,需要先用 Excel 打开mixed_galt.txt,使用“分列工具”按照空格分开。使用数据工具中的“去除重复项”。你就可以得到这些序列。理论上这样子筛选出来的基因id不会比网站多太多,用 Araport11 的文件搜索是差不多的。但是用 TAIR10 的文件就会搜出很多结果。一定程度上我建议大家单纯参考网站给的搜索结果就好了。不需要再用 linux 抓取。
获取了基因id之后,我们就可以开启TBtools来提取我们需要的蛋白质序列了。
在 TBtools 中,所有跟腱都可以拖入那些条条框框中,你也可以自己手动改输入输出路径。
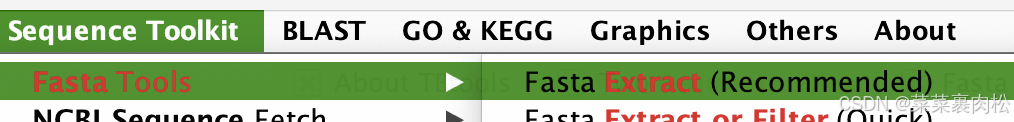
Input 放 拟南芥pep文件,output复制粘贴同样的路径,把文件名改成 ath.rawgalt.pep,ID list 粘贴上我们刚刚拿到的基因id。注意如果是 linux grep 出来的,用Excel 改写一下。只保留id名称。
#如果是使用终端呢?
需要使用到 seqkit,可以用 conda 安装
#安装seqkit
conda install bioconda::seqkit
#提取蛋白质序列,-f是file,-o是output
seqkit grep -f ath.galt.id.txt ath.pep -o ath.rawgalt.pep这里我们还需要自己编写一个python文件来取出多余的id信息,从而简化id。
#我们需要使用一个新的指令,vim或者vi
vim id.simplify.py
#你会进入一个空的文件里,这你是刚刚创建的id.simplify.py
#首先点击键盘上的i,相当于insert。再复制下面的python脚本,粘贴进去(command+v)
#再点击键盘上的esc退出insert模式,输入:wq,w就是write写入,q就是quit退出。回车后,你就写完了这个python文件了。
#简化id
python3 id.simplify.py ath.rawgalt.pep ath.galt.pep
#输出文件 ath.galt.pep 的基因id就是被简化过的了
#python脚本
——————————————————————————————————————————————
#假设我们输入的ath.rawgalt.txt,输出ath.galt.pep
from sys import argv
pepfile_name = argv[1]
outfile_name = argv[2]
pepfile = open(pepfile_name,'r')
outfile = open(outfile_name,'w')
for line in pepfile:
if line.startswith('>'):
line=line.split(" ")
id = line[0]
print(id, file = outfile)
else:
print(line, end = "", file = outfile)
pepfile.close()
outfile.close()
——————————————————————————————————————————————二、Pfam获取此家族 hmm 文件
Pfam现在和InterPro在一起,会直接跳转,这导致很多老教程都失效了。
点击KEYWORD SEARCH,输入你的家族基因名称,直接GO。

先别慌,看起来结果很多呀!实际上注意中间这列 Source Database。我们找到所有 Pfam 的就好了。

注意到上面两个都是和N端有关系的,最下面这个PF01762比较适合我们。同时如果你能使用维基百科的话,维基百科上有各种基因家族的Pfam号。这个也可以作为参考。确定是PF01762后,点击进入。
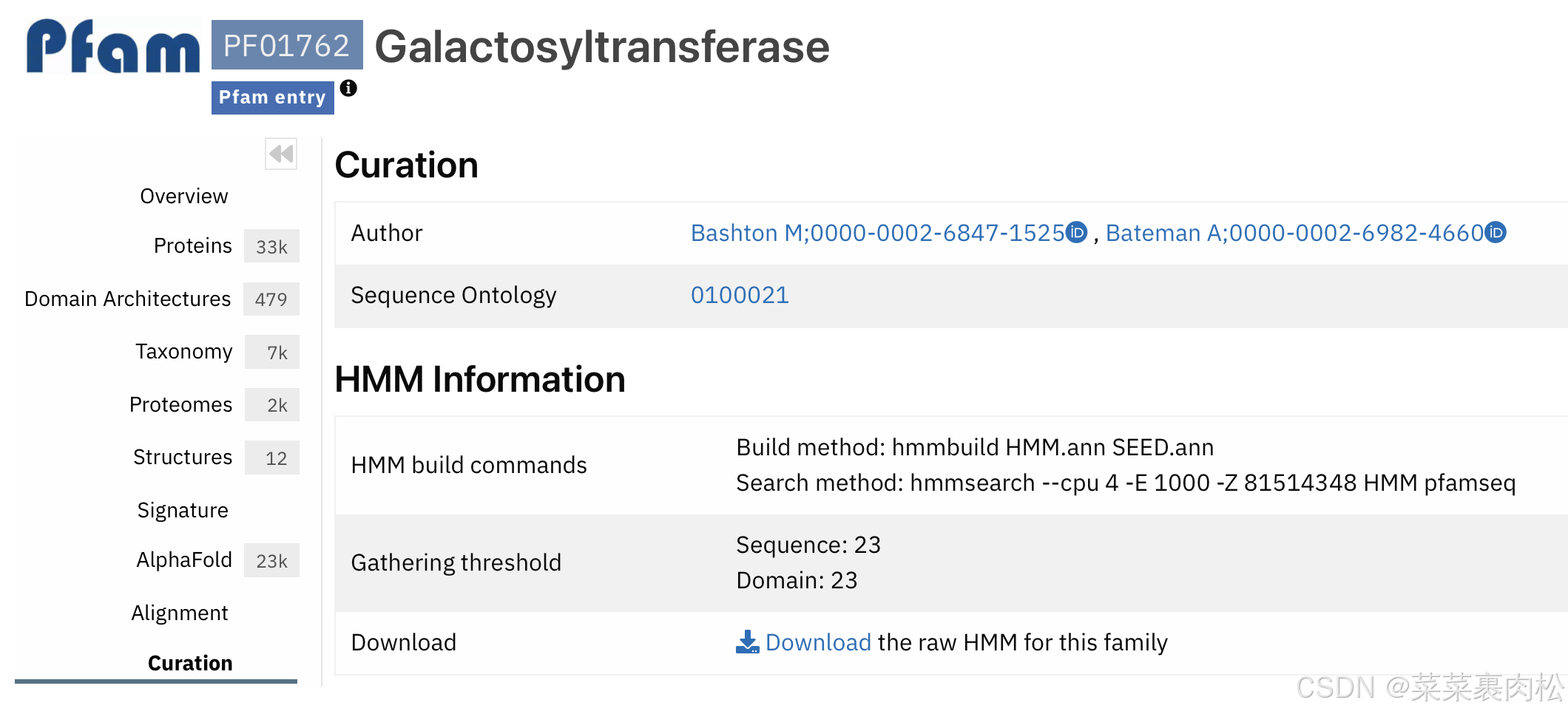
进入到我们的 Curation 栏后,直接Download。这就是我们的 hmm 文件啦。用于后续的 Hmmsearch。
三、BLAST 比对
我们在一中获取了ath.rawgalt.pep。为什么是raw因为id还太长了,要剪一下。
同样的办法,你可以命名为ath.cooked.galt.pep。也可以是别的,无所谓。只要你知道这个文件是干什么的就好了。
然后就是我们最重要的BLAST ALIGNMENT


请注意,右边的Outfmt一定要改为Table才好。Query 是输入序列,用拟南芥的。Subject 用的是库,用你的物种总蛋白质文件。确定好输出路径就可以运行了。
表格的结果为
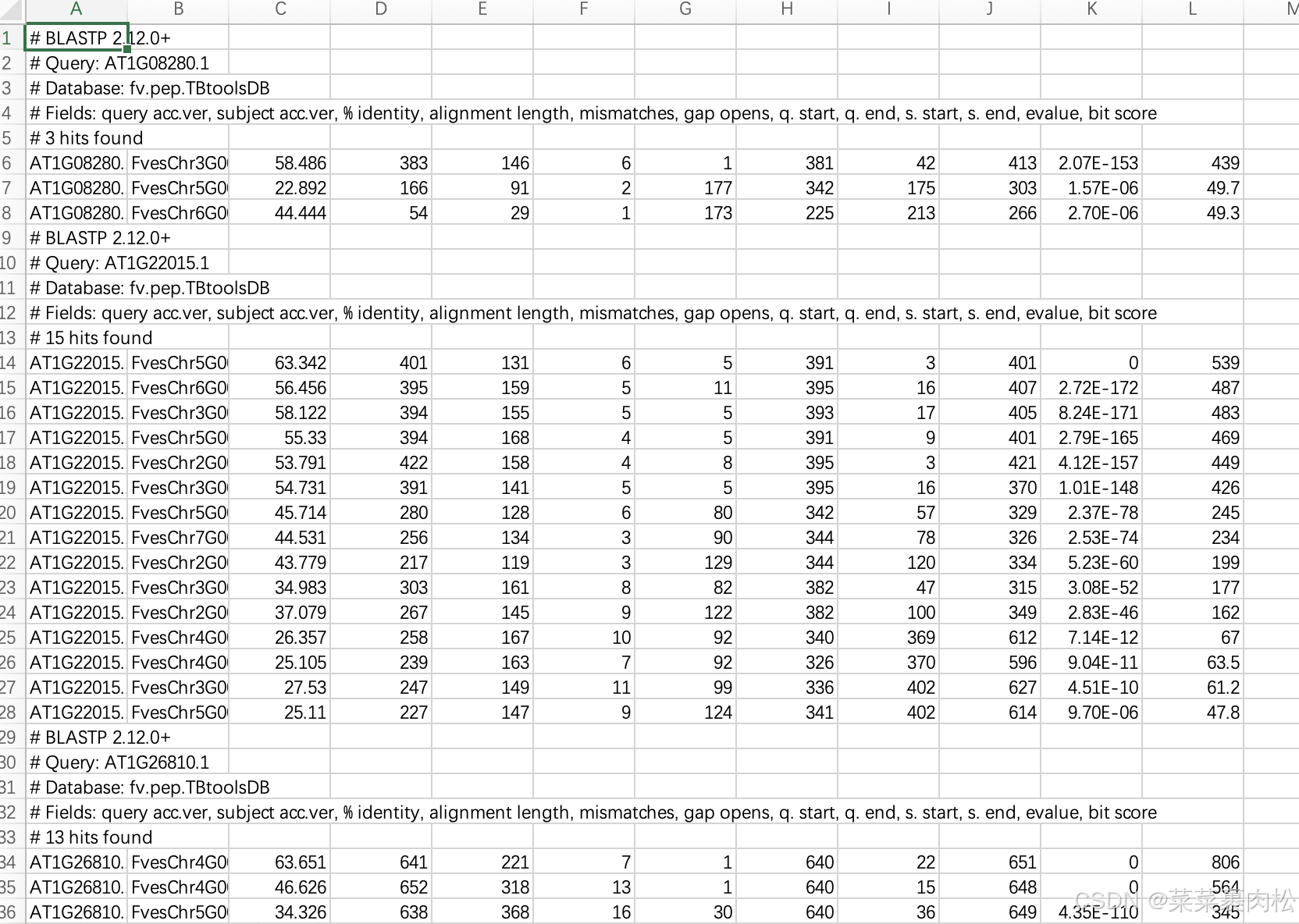
实际上很清楚,每一个 query 都有好几个结果,我们只保存B列的基因id,剩下的全部删除,重新排序,去除重复项就好了。
#如果是使用终端呢?
首先需要你安装BLAST程序,一种是下载安装包,一种是使用conda安装。
conda是一个很厉害的安装程序,一般使用 conda install program_name 这一行指令就可以直接安装了。所以还得先安装 conda 再安装 blast。这边的建议是先安装 miniconda,就可以使用miniconda 来安装 blast。
而且如果想用终端完成全部的工作,conda 是必须的工具。
一般可以直接用conda install bioconda::blast,conda install bioconda::hmmer来直接安装。如果报错,则在anaconda官网找这个包。
有了BLAST程序以后我们就可以使用这简简单单三行代码去实现上面的功能了,关键在于一下代码可以直接复制粘贴就运行。不需要鼠标动来动去。如果我们需要BLAST出10个物种的GALT蛋白质,绝对是终端法更方便。
#安装BLAST
conda install bioconda::blast
#subject建库
makeblastdb -dbtype prot -in fv.pep -input_type fasta -parse_seqids -out fv.blastdb
#query参与blast比对,准确来说是blastp(蛋白质版),还有一个是blastn(核苷酸版)
blastp -query ath.galt.pep -db fv.blastdb -out fv.blastp.xls -task blastp
#处理结果
grep '^>' fv.blastp.xls| awk '{print $1}' | sed 's/>//' | sort | uniq > fv.blast.list.txt
终端法有没有缺点呢?有,那就是你没有进入到文件里看文件到底怎么样,所以我们需要用到 head 指令来检查我们的文件也没有问题。以下是一个批量操作的示范。
#建库
makeblastdb -dbtype prot -in pbr.pep -input_type fasta -parse_seqids -out pbr.blastdb
makeblastdb -dbtype prot -in md.pep -input_type fasta -parse_seqids -out md.blastdb
makeblastdb -dbtype prot -in sl.pep -input_type fasta -parse_seqids -out sl.blastdb
makeblastdb -dbtype prot -in vv.pep -input_type fasta -parse_seqids -out vv.blastdb
makeblastdb -dbtype prot -in pp.pep -input_type fasta -parse_seqids -out pp.blastdb
makeblastdb -dbtype prot -in fv.pep -input_type fasta -parse_seqids -out fv.blastdb
makeblastdb -dbtype prot -in cs.hpa.pep -input_type fasta -parse_seqids -out cs.blastdb
#blast比对
blastp -query ath.galt.pep -db pbr.blastdb -out pbr.blastp.xls -task blastp
blastp -query ath.galt.pep -db md.blastdb -out md.blastp.xls -task blastp
blastp -query ath.galt.pep -db sl.blastdb -out sl.blastp.xls -task blastp
blastp -query ath.galt.pep -db vv.blastdb -out vv.blastp.xls -task blastp
blastp -query ath.galt.pep -db pp.blastdb -out pp.blastp.xls -task blastp
blastp -query ath.galt.pep -db fv.blastdb -out fv.blastp.xls -task blastp
blastp -query ath.galt.pep -db cs.blastdb -out cs.blastp.xls -task blastp
#处理结果,先抓取带>号的行,只print第一列(就是基因id),然后把每一个id的>号去掉
grep '^>' pbr.blastp.xls| awk '{print $1}' | sed 's/>//' | sort | uniq > pbr.blast.list.txt
grep '^>' md.blastp.xls| awk '{print $1}' | sed 's/>//' | sort | uniq > md.blast.list.txt
grep '^>' sl.blastp.xls| awk '{print $1}' | sed 's/>//' | sort | uniq > sl.blast.list.txt
grep '^>' vv.blastp.xls| awk '{print $1}' | sed 's/>//' | sort | uniq > vv.blast.list.txt
grep '^>' pp.blastp.xls| awk '{print $1}' | sed 's/>//' | sort | uniq > pp.blast.list.txt
grep '^>' fv.blastp.xls| awk '{print $1}' | sed 's/>//' | sort | uniq > fv.blast.list.txt
grep '^>' cs.blastp.xls| awk '{print $1}' | sed 's/>//' | sort | uniq > cs.blast.list.txt
#查看结果
head *.blast.list.txt四、Hmmsearch
前面我们已经有了 hmm 文件。我们还需要编辑一个空txt文本,在里面写上我们的Pfam号,比如PF01762。
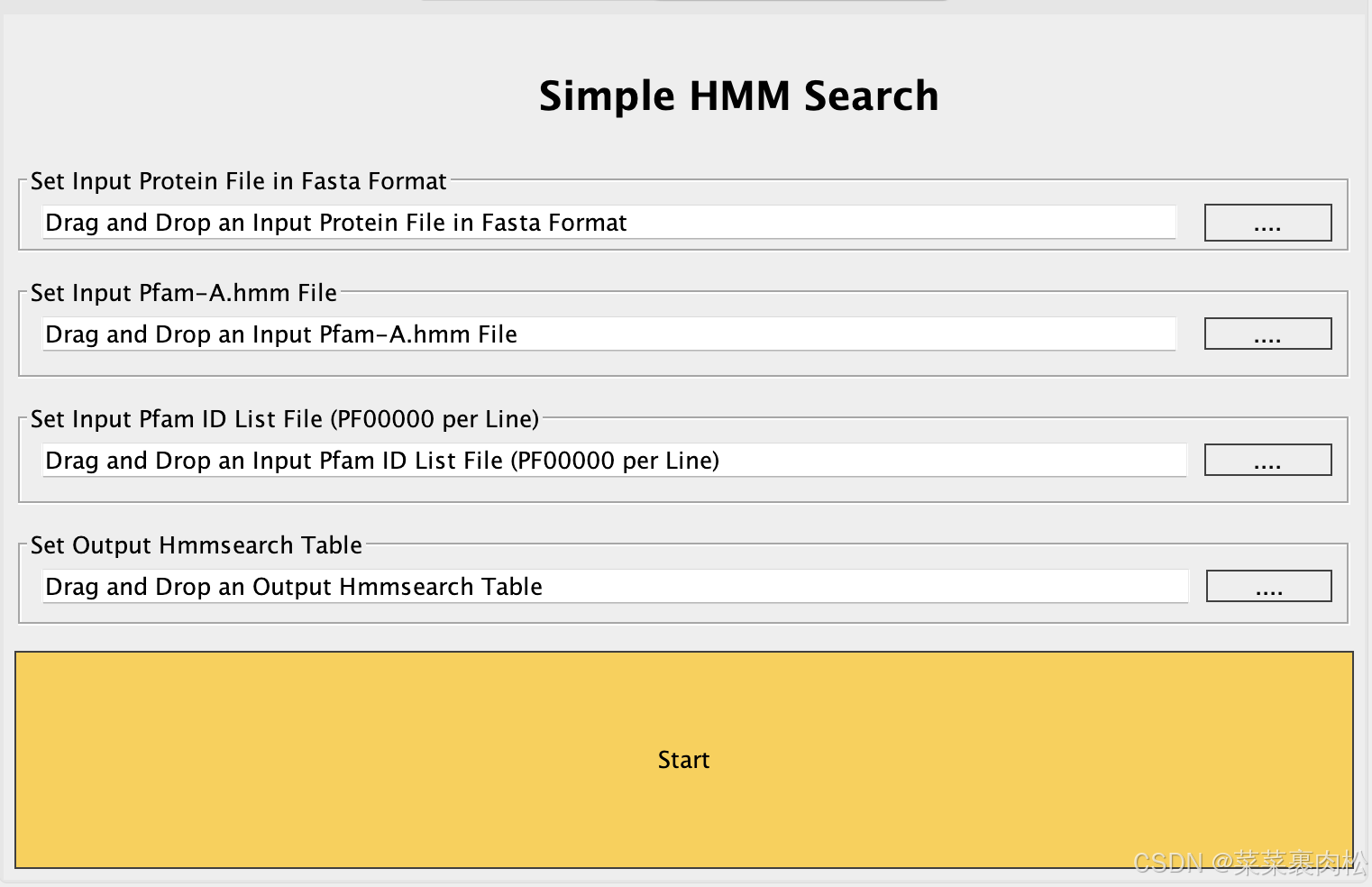
- 物种蛋白质文件
- hmm 文件
- 有 PF 号的 txt 文件
- 设置输出路径和文件名
与BLAST结果的处理方式一样,只保留基因id,排序后取出重复项。
#如果是使用终端呢?
以下也是一个批量操作的示范
#安装hmmer3.4
conda install bioconda::hmmer
#hmm搜索
hmmsearch PF01762.hmm-3 pbr.pep > pbr.hmm.xls
hmmsearch PF01762.hmm-3 md.pep > md.hmm.xls
hmmsearch PF01762.hmm-3 sl.pep > sl.hmm.xls
hmmsearch PF01762.hmm-3 vv.pep > vv.hmm.xls
hmmsearch PF01762.hmm-3 pp.pep > pp.hmm.xls
hmmsearch PF01762.hmm-3 fv.pep > fv.hmm.xls
hmmsearch PF01762.hmm-3 cs.hpa.pep > cs.hmm.xls
#处理结果
grep '^>' pbr.hmm.xls | awk '{print $2}' > pbr.hmm.list.txt
grep '^>' md.hmm.xls | awk '{print $2}' > md.hmm.list.txt
grep '^>' sl.hmm.xls | awk '{print $2}' > sl.hmm.list.txt
grep '^>' vv.hmm.xls | awk '{print $2}' > vv.hmm.list.txt
grep '^>' pp.hmm.xls | awk '{print $2}' > pp.hmm.list.txt
grep '^>' fv.hmm.xls | awk '{print $2}' > fv.hmm.list.txt
grep '^>' cs.hmm.xls | awk '{print $2}' > cs.hmm.list.txt五、Venn图得交集
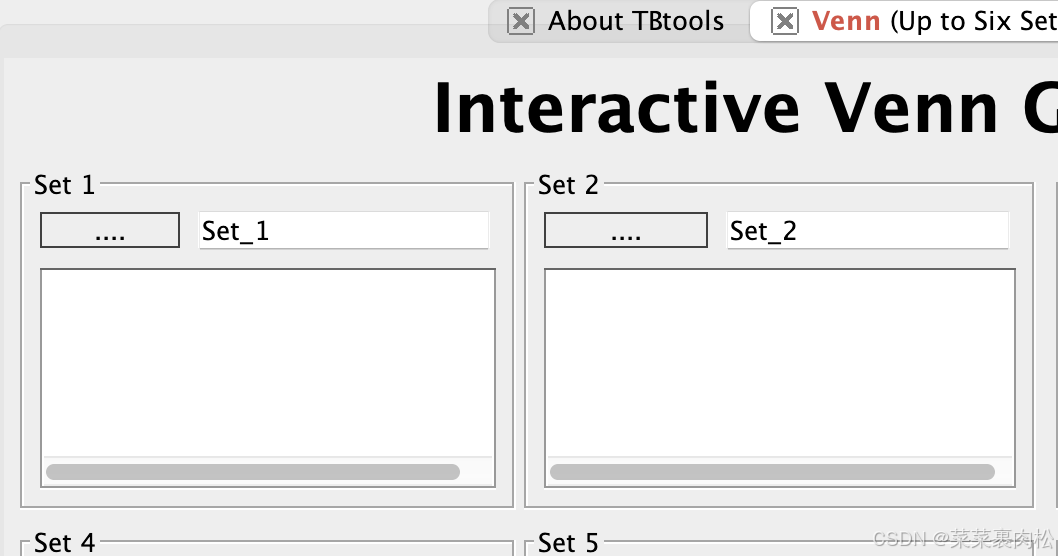
我们只用得上这两个 Set。把之前处理的BLAST结果和hmm结果放进去,获得交集的基因id,就是我们的候选基因啦!
#如果是使用终端呢?
#求交集
sort pbr.hmm.list.txt pbr.blast.list.txt | uniq -d > pbr.galt.id.txt
sort md.hmm.list.txt md.blast.list.txt | uniq -d > md.galt.id.txt
sort sl.hmm.list.txt sl.blast.list.txt | uniq -d > sl.galt.id.txt
sort vv.hmm.list.txt vv.blast.list.txt | uniq -d > vv.galt.id.txt
sort pp.hmm.list.txt pp.blast.list.txt | uniq -d > pp.galt.id.txt
sort fv.hmm.list.txt fv.blast.list.txt | uniq -d > fv.galt.id.txt
sort cs.hmm.list.txt cs.blast.list.txt | uniq -d > cs.galt.id.txt
六、获取最终的候选基因序列并简化id
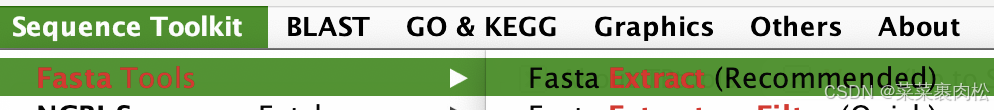
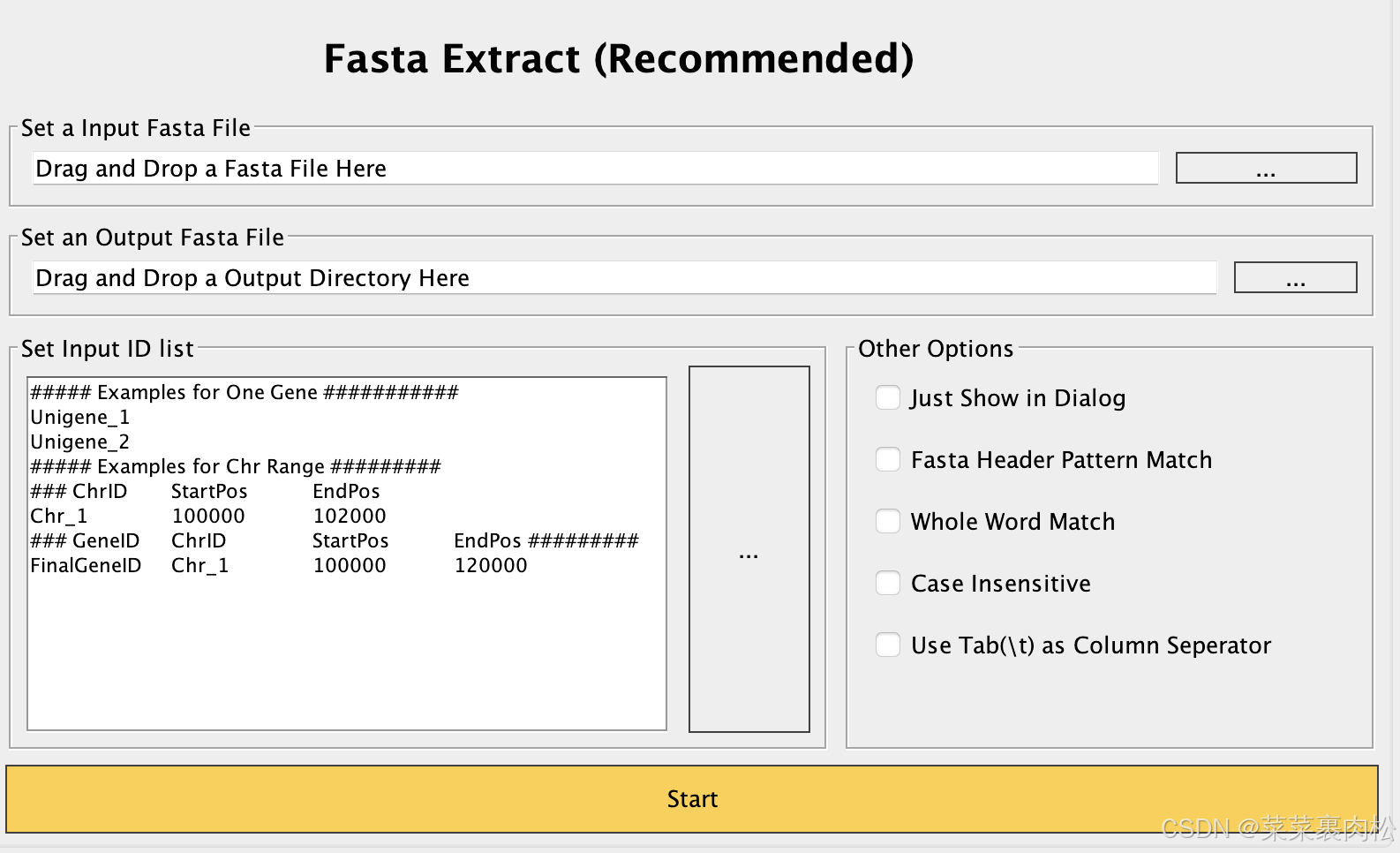
看了这么久,你应该知道先放蛋白质文件,再写输出路径,再把基因id贴进去。就能得到所需要的蛋白质序列。下面简化id的操作也是一样简单的。
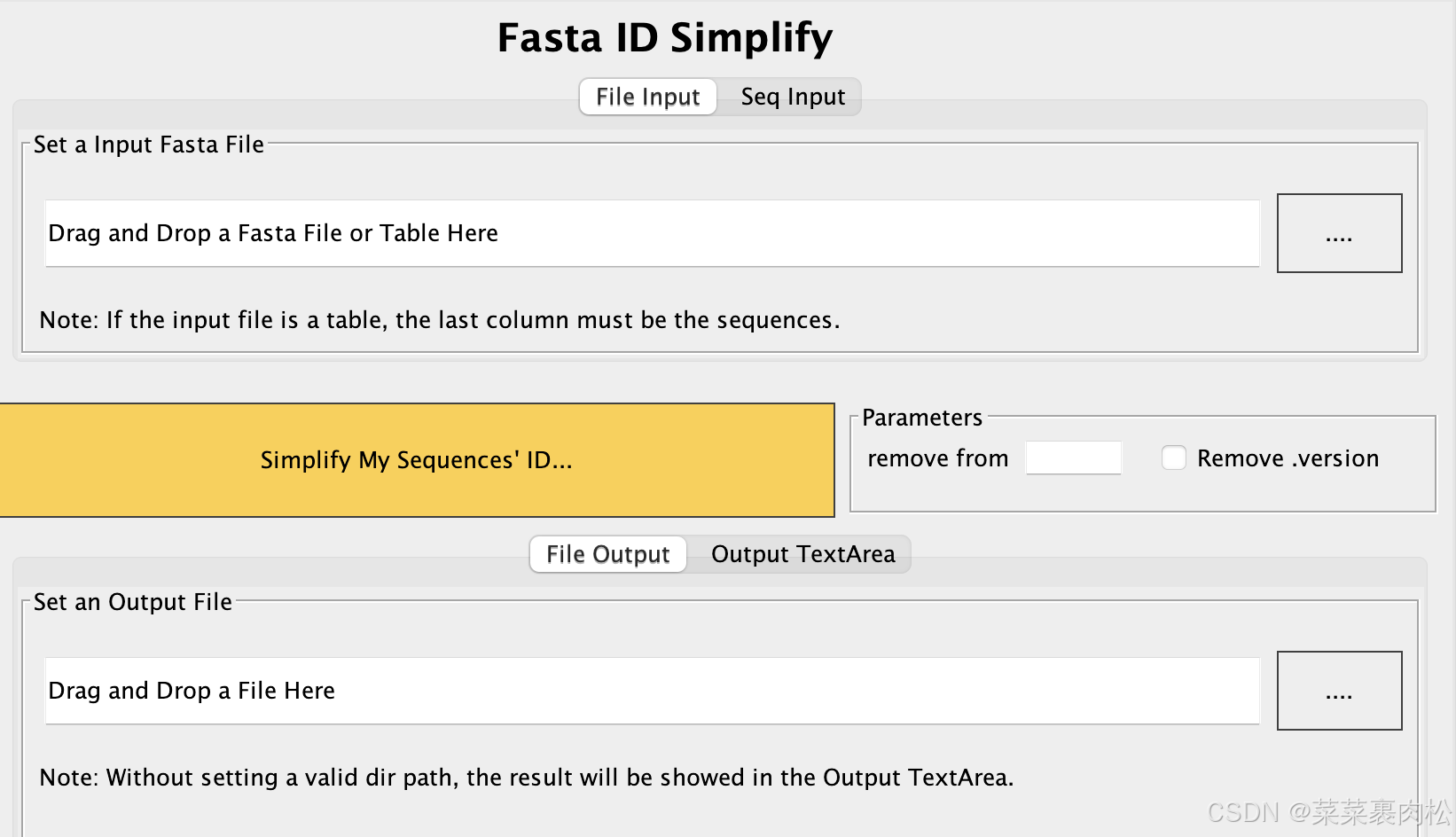
#如果是使用终端呢?
#和我们提取拟南芥序列是一样的
seqkit grep -f pbr.galt.id.txt pbr.pep -o pbr.rawgalt.pep
seqkit grep -f md.galt.id.txt md.pep -o md.rawgalt.pep
seqkit grep -f sl.galt.id.txt sl.pep -o sl.rawgalt.pep
seqkit grep -f vv.galt.id.txt vv.pep -o vv.rawgalt.pep
seqkit grep -f pp.galt.id.txt pp.pep -o pp.rawgalt.pep
seqkit grep -f fv.galt.id.txt fv.pep -o fv.rawgalt.pep
seqkit grep -f cs.galt.id.txt cs.pep -o cs.rawgalt.pep(补充)终端全部的指令
你要懒得话,把我这个复制到你的txt文本里,把文件名改成你自己的,再直接放进终端里粘贴回车也可以了。
#先确保你创建了x64架构的虚拟环境
conda activate x64env
conda install bioconda::blast
conda install bioconda::hmmer
conda install bioconda::seqkit
指令生成python文件 generate_command.py
#generate_command.py
from sys import argv
if argv[1] == '-h':
print("\033[1;31mYou should type\033[0m python3 generate_command.py sp_list gene_name hmmfile_name > your_linux_command")
else:
splist = argv[1]
gene = argv[2]
hmmfile = argv[3]
def generate(sp,gene,hmmfile):
print(f"makeblastdb -dbtype prot -in {sp}.pep -input_type fasta -parse_seqids -out {sp}.blastdb")
print(f"blastp -query ath.{gene}.pep -db {sp}.blastdb -out {sp}.blastp.xls -task blastp")
print(f"grep '^>' {sp}.blastp.xls|","awk '{print $1}'",f"| sed 's/>//' | sort | uniq > {sp}.blast.list.txt")
print(f"hmmsearch {hmmfile} {sp}.pep > {sp}.hmm.xls")
print(f"grep '^>' {sp}.hmm.xls |"," awk '{print $2}'",f" > {sp}.hmm.list.txt")
print(f"sort {sp}.hmm.list.txt {sp}.blast.list.txt | uniq -d > {sp}.{gene}.id.txt")
print(f"python3 fa_grep.py {sp}.{gene}.id.txt {sp}.pep > {sp}.{gene}.pep")
data = []
for line in open(splist,'r'):
data.append(line.strip())
for item in data:
generate(item,gene,hmmfile)python3 generate_command.py id kunkun PF000250.hmm > out.command
#也可以使用 python3 generate_command.py -h
#你就会得到 You should type python3 generate_command.py sp_list gene_name hmmfile_name > your_linux_command
# id 文件内容为
pbr
md
sl
pp
vv
ath
tt
or
fv
# out.command 结果为
makeblastdb -dbtype prot -in pbr.pep -input_type fasta -parse_seqids -out pbr.blastdb
blastp -query ath.kunkun.pep -db pbr.blastdb -out pbr.blastp.xls -task blastp
grep '^>' pbr.blastp.xls| awk '{print $1}' | sed 's/>//' | sort | uniq > pbr.blast.list.txt
hmmsearch PF000250.hmm pbr.pep > pbr.hmm.xls
grep '^>' pbr.hmm.xls | awk '{print $2}' > pbr.hmm.list.txt
sort pbr.hmm.list.txt pbr.blast.list.txt | uniq -d > pbr.kunkun.id.txt
python3 fa_grep.py pbr.kunkun.id.txt pbr.pep > pbr.kunkun.pep
makeblastdb -dbtype prot -in md.pep -input_type fasta -parse_seqids -out md.blastdb
blastp -query ath.kunkun.pep -db md.blastdb -out md.blastp.xls -task blastp
grep '^>' md.blastp.xls| awk '{print $1}' | sed 's/>//' | sort | uniq > md.blast.list.txt
hmmsearch PF000250.hmm md.pep > md.hmm.xls
grep '^>' md.hmm.xls | awk '{print $2}' > md.hmm.list.txt
sort md.hmm.list.txt md.blast.list.txt | uniq -d > md.kunkun.id.txt
python3 fa_grep.py md.kunkun.id.txt md.pep > md.kunkun.pep
makeblastdb -dbtype prot -in sl.pep -input_type fasta -parse_seqids -out sl.blastdb
blastp -query ath.kunkun.pep -db sl.blastdb -out sl.blastp.xls -task blastp
grep '^>' sl.blastp.xls| awk '{print $1}' | sed 's/>//' | sort | uniq > sl.blast.list.txt
hmmsearch PF000250.hmm sl.pep > sl.hmm.xls
grep '^>' sl.hmm.xls | awk '{print $2}' > sl.hmm.list.txt
sort sl.hmm.list.txt sl.blast.list.txt | uniq -d > sl.kunkun.id.txt
python3 fa_grep.py sl.kunkun.id.txt sl.pep > sl.kunkun.pep
makeblastdb -dbtype prot -in pp.pep -input_type fasta -parse_seqids -out pp.blastdb
blastp -query ath.kunkun.pep -db pp.blastdb -out pp.blastp.xls -task blastp
grep '^>' pp.blastp.xls| awk '{print $1}' | sed 's/>//' | sort | uniq > pp.blast.list.txt
hmmsearch PF000250.hmm pp.pep > pp.hmm.xls
grep '^>' pp.hmm.xls | awk '{print $2}' > pp.hmm.list.txt
sort pp.hmm.list.txt pp.blast.list.txt | uniq -d > pp.kunkun.id.txt
python3 fa_grep.py pp.kunkun.id.txt pp.pep > pp.kunkun.pep
makeblastdb -dbtype prot -in vv.pep -input_type fasta -parse_seqids -out vv.blastdb
blastp -query ath.kunkun.pep -db vv.blastdb -out vv.blastp.xls -task blastp
grep '^>' vv.blastp.xls| awk '{print $1}' | sed 's/>//' | sort | uniq > vv.blast.list.txt
hmmsearch PF000250.hmm vv.pep > vv.hmm.xls
grep '^>' vv.hmm.xls | awk '{print $2}' > vv.hmm.list.txt
sort vv.hmm.list.txt vv.blast.list.txt | uniq -d > vv.kunkun.id.txt
python3 fa_grep.py vv.kunkun.id.txt vv.pep > vv.kunkun.pep
makeblastdb -dbtype prot -in ath.pep -input_type fasta -parse_seqids -out ath.blastdb
blastp -query ath.kunkun.pep -db ath.blastdb -out ath.blastp.xls -task blastp
grep '^>' ath.blastp.xls| awk '{print $1}' | sed 's/>//' | sort | uniq > ath.blast.list.txt
hmmsearch PF000250.hmm ath.pep > ath.hmm.xls
grep '^>' ath.hmm.xls | awk '{print $2}' > ath.hmm.list.txt
sort ath.hmm.list.txt ath.blast.list.txt | uniq -d > ath.kunkun.id.txt
python3 fa_grep.py ath.kunkun.id.txt ath.pep > ath.kunkun.pep
makeblastdb -dbtype prot -in tt.pep -input_type fasta -parse_seqids -out tt.blastdb
blastp -query ath.kunkun.pep -db tt.blastdb -out tt.blastp.xls -task blastp
grep '^>' tt.blastp.xls| awk '{print $1}' | sed 's/>//' | sort | uniq > tt.blast.list.txt
hmmsearch PF000250.hmm tt.pep > tt.hmm.xls
grep '^>' tt.hmm.xls | awk '{print $2}' > tt.hmm.list.txt
sort tt.hmm.list.txt tt.blast.list.txt | uniq -d > tt.kunkun.id.txt
python3 fa_grep.py tt.kunkun.id.txt tt.pep > tt.kunkun.pep
makeblastdb -dbtype prot -in or.pep -input_type fasta -parse_seqids -out or.blastdb
blastp -query ath.kunkun.pep -db or.blastdb -out or.blastp.xls -task blastp
grep '^>' or.blastp.xls| awk '{print $1}' | sed 's/>//' | sort | uniq > or.blast.list.txt
hmmsearch PF000250.hmm or.pep > or.hmm.xls
grep '^>' or.hmm.xls | awk '{print $2}' > or.hmm.list.txt
sort or.hmm.list.txt or.blast.list.txt | uniq -d > or.kunkun.id.txt
python3 fa_grep.py or.kunkun.id.txt or.pep > or.kunkun.pep
makeblastdb -dbtype prot -in fv.pep -input_type fasta -parse_seqids -out fv.blastdb
blastp -query ath.kunkun.pep -db fv.blastdb -out fv.blastp.xls -task blastp
grep '^>' fv.blastp.xls| awk '{print $1}' | sed 's/>//' | sort | uniq > fv.blast.list.txt
hmmsearch PF000250.hmm fv.pep > fv.hmm.xls
grep '^>' fv.hmm.xls | awk '{print $2}' > fv.hmm.list.txt
sort fv.hmm.list.txt fv.blast.list.txt | uniq -d > fv.kunkun.id.txt
python3 fa_grep.py fv.kunkun.id.txt fv.pep > fv.kunkun.pep
fa_grep.py 脚本
from Bio import SeqIO
from sys import argv
if argv[1] == '-h':
print("\033[1;31mYou should type\033[0m python3 fa_grep.py id_file pep_file > result_file")
else:
idfile_name = argv[1]
fullfile_name = argv[2]
ls = {}
for fa in SeqIO.parse(fullfile_name, 'fasta'):
ls[fa.id.split(' ')[0]] = fa.seq
idfile = open(idfile_name,'r')
for idline in idfile:
id = idline.strip()
print(f'>{id}')
seq = ls[id.strip()]
print(seq)
idfile.close()
















 5万+
5万+

 被折叠的 条评论
为什么被折叠?
被折叠的 条评论
为什么被折叠?
 到【灌水乐园】发言
到【灌水乐园】发言









