






在科研工作里,构建与美化进化树是必须掌握的技能之一。这篇文章汇总和整理了之前的相关内容,依照文章中的脉络,基本上能够在大多数情形下满足构建进化树的需求。本文将从测序数据到构建系统发育树需要经过这4个步骤进行阐述:①数据收集与预处理②多序列比对③选择适合的建树方法④系统发育树的评估与美化。
数据准备与预处理
数据收集与预处理是基因组测序数据分析的关键步骤,主要包括去除低质量序列、去除接头序列、过滤污染序列等,以提高数据的整体质量。
如何选择合适的分子序列
在构建系统发育树之前,首先需要从海量的生物信息数据中挑选出适合的分子序列。常见的分子序列包括DNA序列和蛋白质序列。
• DNA序列:DNA分子由四种核苷酸(A、T、G、C)排列组成,DNA序列就是这些核苷酸的排列次序。DNA序列具有高变异性和丰富的遗传信息,适合用于亲缘关系较远的物种间的系统发育分析。
• 蛋白质序列:蛋白质的一级结构由20种氨基酸线性排列构成,蛋白序列就是这些氨基酸的排列次序。蛋白质序列相对保守,适合用于亲缘关系较近的物种间的系统发育分析。
选择分子序列时,首先要明确研究目的。若想探究物种间大框架的亲缘关系,那就选保守性高的基因序列,比如 16S rRNA 基因,在原核生物系统发育研究中就经常会用到。要是关注物种近期的进化,或者种群动态,那就选变异性高的基因或者非编码区序列。同时,还得考虑物种特性。在植物研究中,叶绿体基因相对保守,非常适合用来分析植物类群的系统发育。而在动物研究里,线粒体基因进化速率快,常用于动物种群层面的研究。
• 注意事项:
• 确保所选序列的质量,避免包含过多噪声或错误。
• 所选序列应能代表目标物种的遗传特征,确保分析结果的可靠性。
• 选的基因进化速率要适中,太慢信息不够,太快又容易出错。
• 确保基因在不同物种里既有保守性又有变异性,这样才能区分不同物种。
• 基因长度和覆盖范围也很重要,太短信息量不够。
数据收集
• 从测序平台获取下机数据,这些数据通常是DNA、RNA或蛋白质的原始序列数据。
• 可以从公共数据库(如NCBI、Ensembl等)获取额外的序列数据,以丰富数据集。
质量控制与预处理
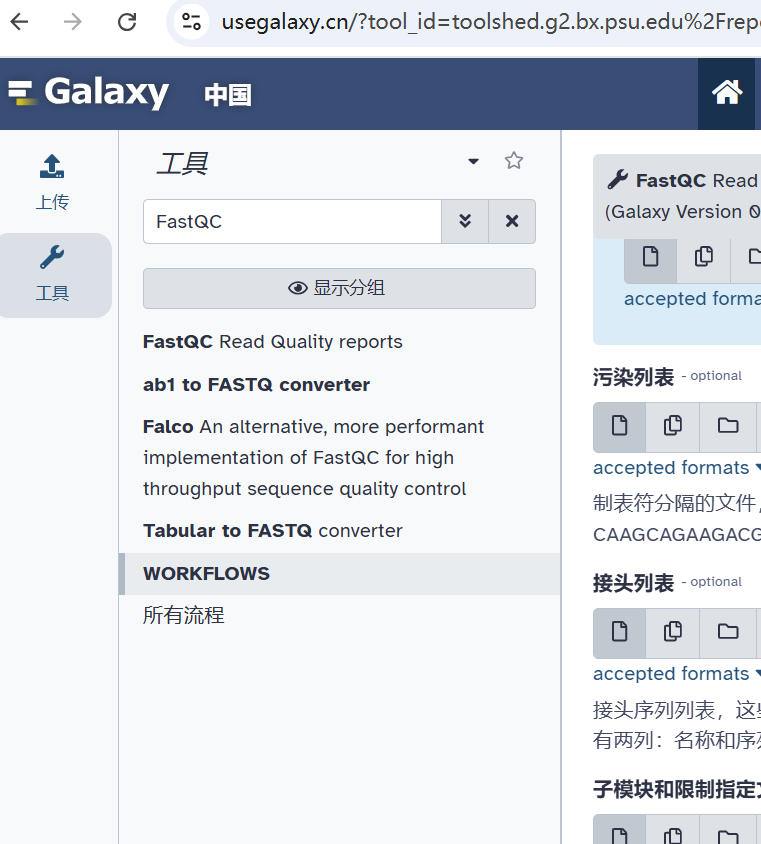
• FastQC
用于评估测序数据的质量,提供碱基质量分布、GC含量分布、序列长度分布等统计信息。
详情参考文章:生信软件,就是赢家通吃:最佳FASTQ质控软件
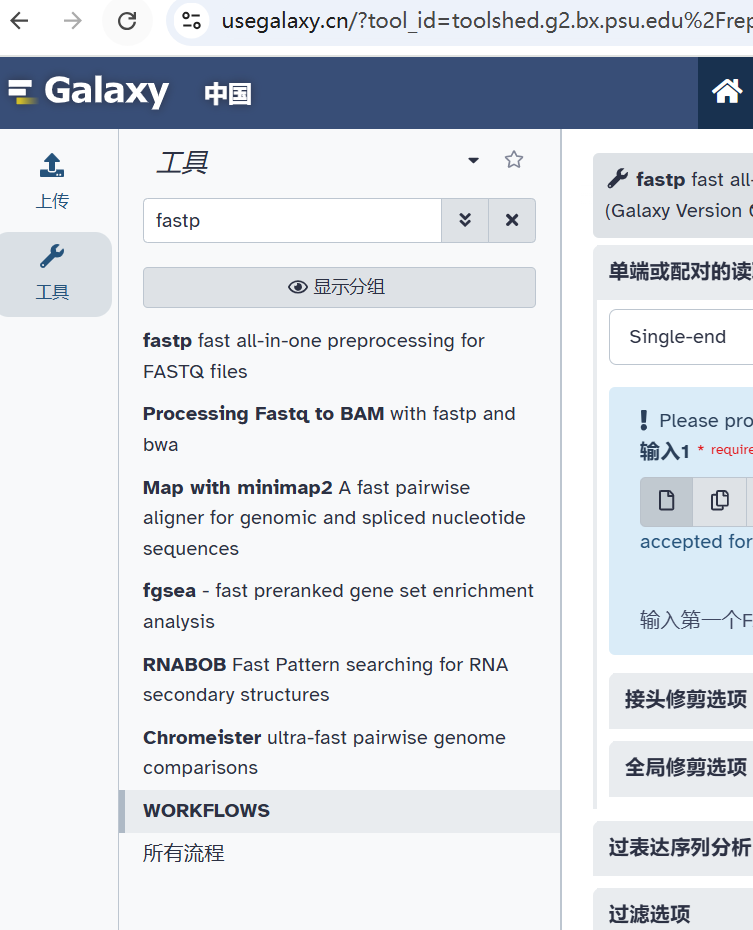
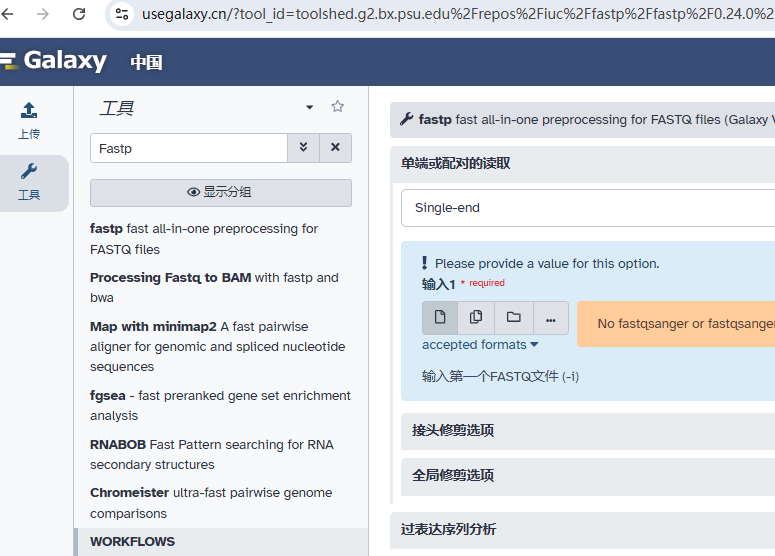
• fastp
一个高效、快速的通用型序列数据质控工具,支持多种质控功能,如低质量碱基修剪、去除接头、过滤低质量读段等。
详情参考文章:都2025年了,谁还不会下一代测序(NGS)数据质控(一)
• Trimmomatic
用于去除低质量序列和接头序列,支持多种参数配置,如设定质量阈值、最小序列长度等。
详情参考文章:都2025年了,谁还不会下一代测序(NGS)数据质控(三)
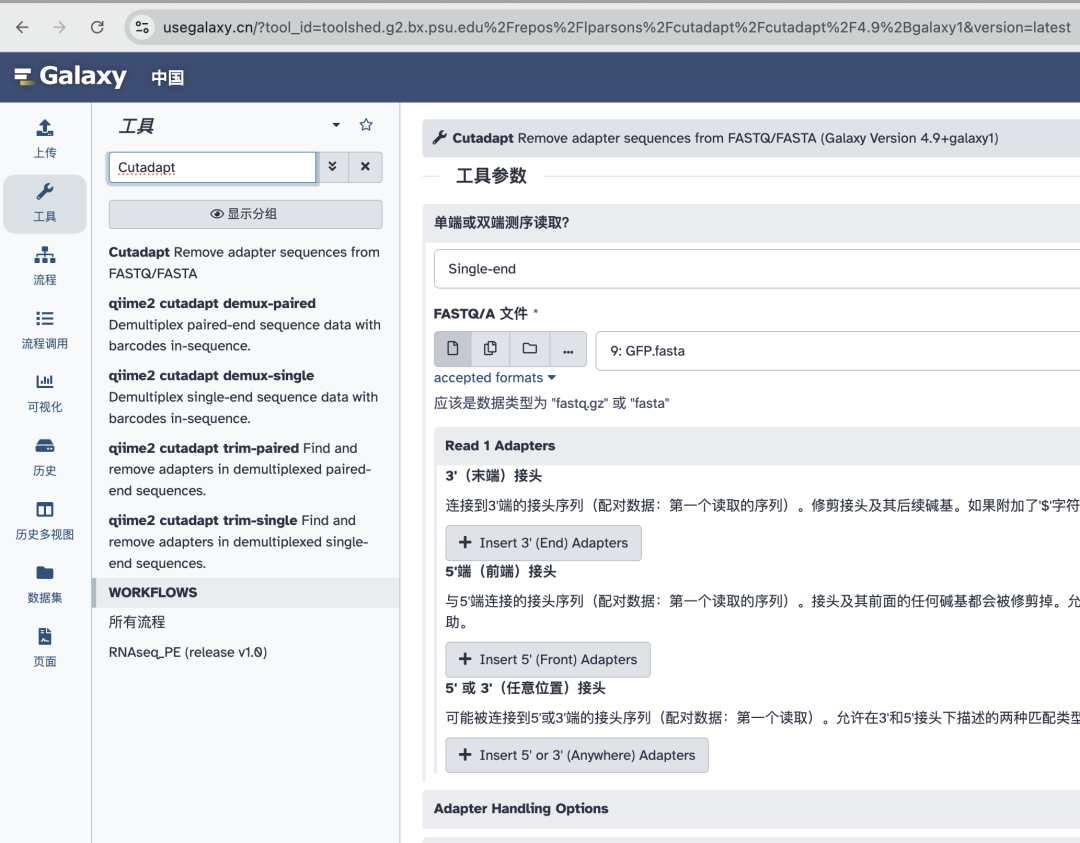
• Cutadapt
专门用于去除接头序列,提高比对的准确性。
详情参考文章:都2025年了,谁还不会下一代测序(NGS)数据质控(四)
• Trim Galore
是一个专注于去除接头序列和低质量碱基的工具,特别适合于 Illumina 测序数据。
详情参考文章:都2025年了,谁还不会下一代测序(NGS)数据质控(二)
• BWA 和 Bowtie2
用于序列比对,将测序读段比对到参考基因组上。
多序列比对
多序列比对软件有哪些,应该怎么选
多序列比对是构建系统发育树的关键步骤之一,通过比对不同物种的分子序列,找出它们之间的相似性和差异性。常用的多序列比对软件包括:
Clustal
老牌软件,操作简单,适合小数据集。基于渐进比对的多序列比对工具,有适用于多种操作平台的版本,如ClustalW和ClustalX。ClustalW速度较慢,但比对结果较为可靠。
MAFFT
针对大数据集(超千条,万条以上序列)的多序列比对软件。处理大量序列时表现出色,速度快,功能强大,能快速处理复杂的任务。如果你不会命令行操作,觉得在本地安装和配置MAFF太麻烦,可以在Galaxy生信云平台上(usegalaxy.cn)运行它来进行多序列比对。
了解详情请参考文章:多序列比对工具,我曾经最爱这一款
Muscle
平衡了速度和准确性,适合中等规模的数据集。其在速度和精度上都优于ClustalW,尤其是在处理一些长度适中,但是有部分序列相似性不是很高的情况。你还可以在Galaxy生信云平台上(usegalaxy.cn)一键运行Muscle来快速进行多序列比对,无需任何安装及配置。
了解详情请参考文章:细菌全基因组序列怎么构建系统进化树啊:详细思路解读,一文掌握
BLAST
最常用的短序列比对工具,支持核酸和蛋白的双序列比对,还可以在数据库中寻找相似序列。BLAST功能强大,但分析速度较慢,结果不够直观。
BLAT
适用于寻找高相似度的序列,对于DNA序列,要求95%及以上相似且至少25个碱基;对于蛋白序列,要求80%及以上相似且至少20个氨基酸。BLAT在远亲缘物种间的比对精度不够高。
DNAMAN
简单常用的核酸序列和蛋白质序列分析软件,支持多序列比对、序列同源性分析等多种功能,界面友好,占用内存小。
选软件的时候,如果序列数量少、长度短,Clustal 系列就行,操作简单且结果好。如果数据集非常大,比如说要对整个基因组中的很多基因进行比对,那可能Clustal Omega或者MAFFT更合适。如果是中等规模,而且希望比对结果比较精确,MUSCLE是个不错的选择。序列相似性高的话,多数软件都能适用;要是相似性低,MAFFT 和 MUSCLE 适应性更好。另外,如果计算资源充足且是多核处理器,Clustal Omega 和 MAFFT 能利用并行计算,速度更快;要是计算资源有限,MUSCLE 也能应对。
注意事项
• 得了解软件原理和算法,选择合适的比对算法和参数,确保比对的准确性和可靠性。
• 比对结果需要用可视化工具检查,或者用评估指标和工具量化评估比对结果的质量和可靠性。
• 删除或处理比对结果中的gap区域,避免影响最终的建树结果。
选择适合的建树方法及其软件工具
常用建树方法有哪些,具体应该怎么选合适
构建系统发育树的方法多种多样,选择合适的方法对于获得可靠的分析结果至关重要。常用的建树方法包括:
最大似然法(ML)
通过建立进化模型,利用统计模型估计各个分类单位之间的进化距离和树的拓扑结构,选择最大似然值最高的系统树作为最佳解。ML法对数据的要求比较高,适合大数据,适用于有合适分子进化模型的情况,尤其适合远缘物种序列。
• 软件工具:IQ-TREE、RAxML、FastTree、PhyML等。这些软件支持多种进化模型,能够处理大规模数据,提供准确的ML树。如果你不会命令行操作,觉得在本地安装和配置这些工具太麻烦,可以在Galaxy生信云平台上(usegalaxy.cn)运行它们来构建系统发育树。
IQ-TREE
了解IQ-TREE详情参考文章:1分钟构建完美的系统进化树
RAxML
快速且强大的ML建树工具。
了解RAxML详情参考文章:这款10年前发表在生信顶刊上的系统进化树构建软件,还能用吗?
FastTree
针对大数据集(超千条,万条以上序列)系统发育树构建软件。
了解FastTree详情参考文章:FastTree:构建系统进化树,比快更快

PhyML
PHYML以其简单、准确和速度而闻名,适用于较小的数据集,在处理短序列时表现优于其他方法。
了解PhyML详情参考文章:抛砖引玉:生信学习的最高境界
我们可以根据自己的需求和喜好等因素去考量,选最适合自己的那个就是最好的。对于大规模数据集,FastTree和IQTREE是较好的选择;而对于需要高精度结果的研究,则可以选择RaxML或PHYML。
距离法(NJ)
通过测量各个分类单位之间的距离,构建进化距离矩阵,再通过层次聚类等方法构建系统树。常用的距离法有UPGMA和Neighbor-Joining(NJ)等。NJ法适用于近缘物种序列,但对于相似度很低的序列可能会出现长枝吸引(LBA)现象。
• 软件工具:MEGA、PHYLIP等。这些软件提供了多种距离法建树选项,操作简便,适合初学者使用。
贝叶斯推理法(BI)
通过建立贝叶斯统计模型,利用贝叶斯推理计算出各个分类单位之间的进化关系和树的拓扑结构概率分布,选择概率最高的系统树作为最佳解。贝叶斯法通常能获得比ML法更准确的结果,但计算量较大。
• 软件工具:MrBayes、BEAST等。这些软件适用于复杂的进化分析,能够提供详细的贝叶斯后验概率分布结果。
最大简约法(MP)
通过比较各个分类单位之间的特征相似性,选择具有最少进化步骤的系统树作为最佳解。适用于数据较少或计算资源有限的情况。
• 软件工具:PAUP*、TNT等。这些软件专门用于最大简约法的分析,能够提供详细的进化树构建结果。
选择建树方法时,需要根据数据的特性和分析目的来决定。如果数据量很大,ML或者BI可能更适合,不过ML要注意参数调整,BI则需要更多的计算资源。如果是小数据集,MP是个不错的选择。如果序列之间的进化距离比较小,距离法(如NJ)可以快速得到结果。当然,目前最受欢迎的当属最大似然法的几款工具。
注意事项
• 模型选择:根据序列类型(如DNA或蛋白质)选择合适的进化模型。
• 计算资源:贝叶斯法和ML法计算量大,需要足够的计算资源。对于大规模数据集,可能需要使用高性能计算集群。
• 综合评估:不同方法的结果可能存在差异,建议综合评估多个方法的结果来得出最终的系统发育关系。
系统发育树的评估与美化
怎么评估构建的系统发育树
构建完系统发育树后,需要对树进行评估,以确保其准确性和可靠性。常用的评估方法包括:
• Bootstrap检验:这是一个很经典的方法。它通过有放回的抽样方式,从原始数据中多次抽取子数据集,然后在每个子数据集上构建系统发育树,统计每个分支在这些重复构建的树中出现的频率。通过设置较高的bootstrap值(通常大于70,文献中一般为1000),评估进化树的可靠性。Bootstrap值越高,结果越可靠。
• 贝叶斯后验概率:这是贝叶斯推断方法特有的评估方式。它可以给出每个分支的可信度,是基于对整个进化过程的建模和数据的分析得出的。概率越高,说明这个分支的可信度越高。
• Jackknife 检验:和 Bootstrap 类似,不过它是无放回抽样,删除一些位点生成样本再建树,统计相同分支频率得到 Jackknife 支持值,解释和 Bootstrap 支持值差不多。
• 似然比检验(LRT):基于最大似然法,比较两个嵌套的系统发育树模型的似然值,计算似然比,与卡方分布临界值比较,判断两个模型有无显著差异。
注意事项
• 选择合适的评估方法,如引导法或分支支持值
• 评估结果应具有较高的可信度,一般引导值 > 70%或后验概率 > 95%的分支被认为是可靠的。
• 结合多种评估方法,确保结果的可靠性。
进化树美化工具
为了使系统发育树更加直观和美观,可以使用一些进化树美化工具。常用的工具包括:
• iTOL:最受欢迎的在线进化树注释和美化工具,支持多种格式输入和丰富的注释功能。由于iTOL是国外网站,使用时可能需要注意网络状况。
• FigTree:基于Java的进化树可视化软件,小巧易用,可生成高质量的图片,适合用于发表级的图形展示。
• EvolView:由中国科学院北京基因组研究所开发的进化树美化工具,功能类似iTOL,支持一键美化,且因为是国产工具,使用更加流畅。
• tvBOT:交互式国产进化树美化在线工具,支持一键复制进化树的分支标签,整理注释表格非常方便。
• PhyD3:由比利时根特大学开发的在线进化树美化工具,功能强大,但因为是国外网站,使用时可能需要注意网络状况。
• MEGA:不仅可以构建系统发育树,还可以进行美化和编辑,支持多种格式的输出。就像一个多功能的工作台,既能搭建模型又能上色。
• ggtree:适用于需要高度定制化的用户,支持多种图形和统计分析。就像用高级的编程工具来定制模型的每一个细节。
注意事项
• 根据发表需求选择合适的美化工具,确保系统发育树的视觉效果。
• 确保所选美化工具与操作系统和文件格式兼容。
• 保存多种格式的树图,以满足不同用途的需求。
结语
构建系统发育树需要从选择合适的分子序列开始,通过多序列比对确保序列的同源性,选择合适的建树方法构建树,并通过评估确保树的可靠性。使用进化树美化工具可以提升树的可读性和展示效果。每个步骤都有其特定的注意事项,确保数据的准确性和分析的可靠性。希望这篇文章能帮助大家更好地理解和应用这些步骤,如果有任何问题,欢迎在评论区留言讨论!
推荐阅读
一键分析10X单细胞数据(点击图片跳转)
一键分析Bulk转录组数据(点击图片跳转)
简说基因 | 精选文章合辑(点击图片跳转)
生信平台
Galaxy生信云平台(UseGalaxy.cn)致力于降低生信分析门槛,让无专业背景的用户也能轻松分析数据。
• 界面化操作与强大的计算资源。
• 成百上千工具和流程免费使用。
• 丰富的可视化和交互分析工具。
• 强大的数据共享以及协作能力。
联系方式


















 1万+
1万+

 被折叠的 条评论
为什么被折叠?
被折叠的 条评论
为什么被折叠?
 到【灌水乐园】发言
到【灌水乐园】发言









