






点击蓝字 关注我们
多组学数据表征及验证肾透明细胞癌分子亚型,指导精准化疗和免疫治疗

iMeta主页:http://www.imeta.science
研究论文
● 原文链接DOI: https://doi.org/10.1002/imt2.147
● 2023年11月16日,安徽医科大学第一附属医院梁朝朝和香港中文大学(深圳)医学院杜鹃团队在 iMeta 在线发表了题为 “Multi-omics characterization and verification of clear cell renal cell carcinoma molecular subtypes to guide precise chemotherapy and immunotherapy” 的文章。
● 本研究通过多组学数据和可靠的聚类算法提供了透明细胞肾细胞癌 (ccRCC) 分子亚型的新颖见解,希望能为未来ccRCC的精准治疗提供参考。
● 第一作者:孟佳林、江爱民、陆晓凡
● 通讯作者:梁朝朝(liang_chaozhao@ahmu.edu.cn) 、杜鹃(dujuan@cuhk.edu.cn)
● 合作作者:顾迪、葛秦涛、柏素文、周云冬、周骏、郝宗耀、言方荣、王林辉、王海涛
● 主要单位:安徽医科大学第一附属医院;香港中文大学(深圳)医学院;海军军医大学第一附属医院
亮 点
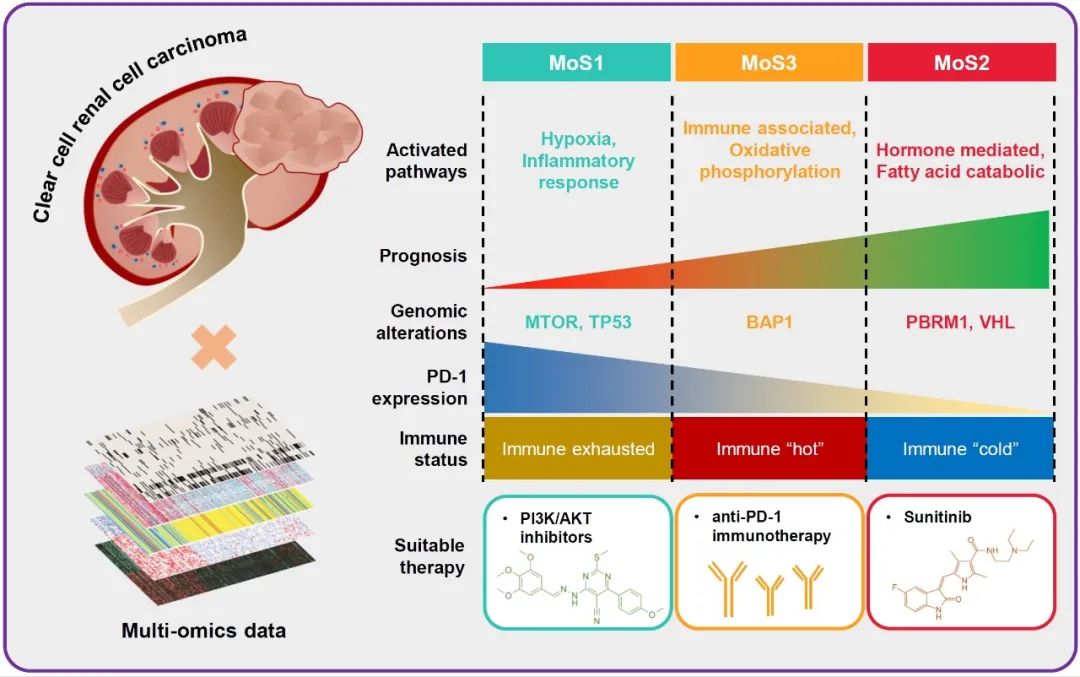
● 基于多组学数据确定了肾透明细胞癌的三种分子亚型,其总生存时间不同,并在外部队列中进行了验证。
● MoS1是免疫衰竭亚型,可受益于PI3K/AKT抑制剂;MoS2是免疫“冷”亚型,更适合舒尼替尼治疗;MoS3是一种免疫“热”亚型,可以受益于抗PD-1免疫治疗。
● SETD2是ccRCC中的肿瘤抑制因子,SETD2的敲除会促进肿瘤细胞增殖、迁移和侵袭。
摘 要
透明细胞肾细胞癌(Clear cell renal cell cancer, ccRCC)是一种具有不同遗传和分子改变的异质性肿瘤,基于多组学分子特征构建ccRCC分类系统刻不容缓,这可以使我们进一步的加深对ccRCC的生物学理解。我们收集了255例ccRCC患者的临床信息、转录组表达谱、拷贝数改变、DNA甲基化和体细胞突变的配对数据,同时,基于我们团队最近开发的R包“MOVICS”进行生物信息分析,通过十种聚类的算法,我们确定了ccRCC患者的多组学亚型 (multi-omics subtype, MoS)。其中,MoS1是免疫衰竭亚型,预后最差,可能是由于免疫微环境衰竭、缺氧特征激活所致,可以受益于PI3K/AKT抑制剂;MoS2属于免疫“冷”亚型,预后良好,VHL和PBRM1突变较多是其突出的分子特征,更适合舒尼替尼治疗。MoS3是免疫“热”亚型,可以从抗PD-1免疫治疗中受益。在外部队列 GSE22541、GSE40435 和 GSE53573 中,我们成功验证了三种MoS的不同分子特征。接受Nivolumab治疗的患者队列帮助我们确认了MoS3 适合抗 PD-1 治疗。E-MTAB-3267队列中的分析结果支持 MoS2 患者对舒尼替尼治疗的反应更好。我们还证实SETD2是ccRCC中的关键抑癌基因,随着肿瘤进展,SETD2蛋白水平降低,敲低SETD2加剧了细胞增殖、迁移和侵袭。总之,我们通过多组学数据和可靠的聚类算法提供了ccRCC分子亚型的新颖见解,希望能为未来ccRCC的精准治疗提供参考。
视频解读
Bilibili:https://www.bilibili.com/video/BV1Rw411n7Wa/
Youtube:https://youtu.be/QSJ6_pvdFfQ
中文翻译、PPT、中/英文视频解读等扩展资料下载
请访问期刊官网:http://www.imeta.science/
全文解读
引 言
在全球范围内,肾癌是所有肿瘤类型中第九位最常见的癌症,其中肾细胞癌(Renal cell cancer, RCC)占此类恶性肿瘤的90%。2018年,有426,800名男性和22,660名女性被诊断出肾细胞癌。据报道,肾细胞癌分别是美国男性和女性中第六和第十大常见肿瘤。全球范围内RCC的发病率差异很大。与亚洲和南美洲相比,北美的发病率较高。即使在一个国家,不同地区的情况也有所不同。例如,萨勒诺每10万人中就有3.6人患有RCC,而意大利东北部地区每10万人中就有9.0人患有RCC。也有证据表明,其发病率会随着年龄的增长而增加,75岁以上的老年人是发病高峰。根据SEER数据库报道,不同种族和性别之间的发病率存在显着差异。非裔美国人的发病率最高,而亚裔/太平洋岛民的发病率仅为其他族裔的一半。无论国家和种族如何,同年龄水平男性的发病率是女性的两倍。对于每个个体来说,仍有许多RCC危险因素已被验证,例如吸烟、高血压和肥胖。近几十年来,在上述影响下,总体发病率呈上升趋势。然而,随着腹部影像学临床应用和综合治疗策略的改良,相对生存率有所提高。根据多种组织病理学特征(例如解剖位置、主要细胞质和染色特征、结构和形态特征),RCC可以分为九个亚型。透明细胞肾细胞癌(Clear cell renal cell cancer, ccRCC)是WHO分类中的主要亚型,占RCC所有亚型的65-70%。ccRCC是一种异质性肿瘤,具有不同的遗传特征和分子改变。尽管RCC的总体生存率不断增加,但ccRCC仍然是最恶性的泌尿系肿瘤之一,每年导致90,000人死亡。考虑到肾细胞癌的高比例和高死亡率,迫切需要一种预测工具和有效的分类系统。目前公认的预后工具基于TNM分期和组织学分化等级。然而,结果是根据腹部影像学和临床病理学确定的,这可能导致大约20%的误差范围。因此,这些工具的临床价值有限,需要研究更有效、更准确的方法。幸运的是,微阵列和高通量测序技术的发展提供了在分子水平上对ccRCC进行亚型分型的机会,这可能有助于更精确的临床指导。该领域已经报道了基于单一mRNA表达或蛋白质特征的多项研究,这研究促进了对ccRCC分子特征和生物学行为之间串扰的理解。然而,这些单一基因组学水平的研究都没有彻底揭示ccRCC分子特征和临床特征的复杂联系。基于此,我们提出了基于多组学的ccRCC分类系统,这可以促进对ccRCC的进一步生物学认识。癌症基因组图谱(Cancer Genome Atlas, TCGA)付出了了大量的努力,收集总结了ccRCC的基因组图谱、转录组图谱和表观遗传学图谱。此外,许多分子改变及其相应的生物过程也已被揭示。这些证据为全面深入探讨ccRCC的分类提供了基础。已经提出了几种ccRCC的多组学整合策略和分类器。宋等人发现FOXM1可以作为ccRCC的新型预后生物标志物,并为分子水平的早期诊断提供基础。越来越多的研究强调肿瘤各种组学之间复杂的相互作用。这种分析方法合并了多个组学的研究结果,能够全面阐明肿瘤分子亚型、预后和药物敏感性。在本研究中,我们纳入了mRNA和长非编码RNA(lncRNA)表达、基因突变、拷贝数改变(Copy number alteration, CNA)和DNA甲基化谱等多组学数据,通过多种聚类算法确认的不同亚型进行了共识集成,建立了一种新颖的综合共识分类,为ccRCC分子特征与生物学行为之间的相关性、生存预测和治疗效果评估提供了更全面、更深入的理解。
结 果
基于多组学数据识别3种亚型
在筛选患者的mRNA、lncRNA表达、CNA、DNA甲基化、基因突变和总生存(Overall survival, OS)结果的所有数据后,我们根据TCGA-KIRC队列中的225名ccRCC患者进行分子亚型的共识分析。根据CPI和Gap,我们最终选择将ccRCC亚型分为3簇。R包“MOVICS”中的十种聚类算法用于将患者分为预设的3种多组学亚型(Multi-omics subtypes, MoS),并最终通过集成共识整合为稳健的分类。轮廓分析的结果还说明了每个簇中样本的中等相似性,MoS1、MoS2和MoS3的轮廓得分分别为0.75、0.50和0.28。图1A显示了上述多组学数据在三种MoS中的分布以及临床病理特征。MoS2含有较多的CNA,但DNA甲基化较少,VHL和PBRM1突变的患者也被分离到MoS2。MoS2中SEMA3G、CHDH、PDK4的mRNA水平最高,而MoS1中DNA甲基化较多,ANLN、APBA2和TMEM132A水平较高。我们还观察到MoS3组中VHL、SETD2和PBRM1的突变较多(图1A)。
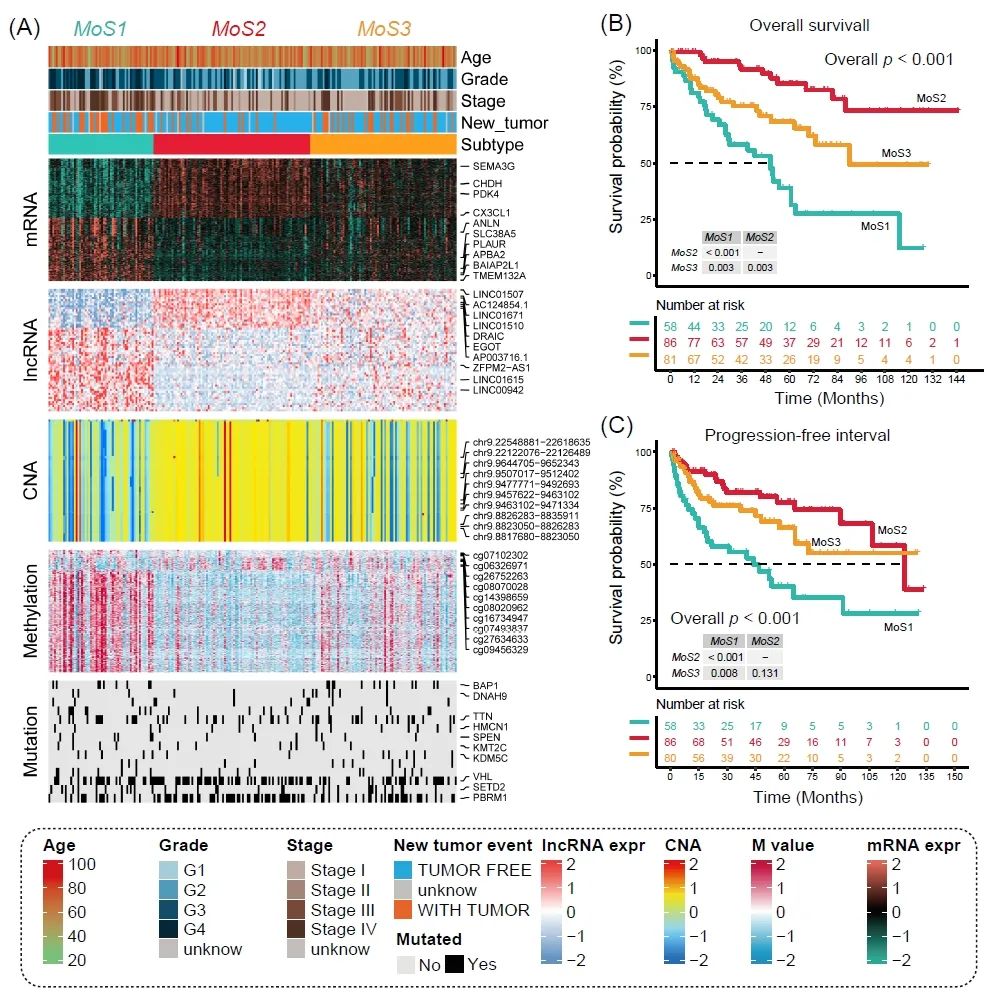
图1. 三个不同MoS簇的识别和不同临床结果的评估
(A) MoS1、MoS2和MoS3亚型的多组学特征。(B) 三种亚型总体生存的Kaplan-Meier图。(C) 三种亚型的无进展间隔的Kaplan-Meier曲线。MoS,多组学亚型;CNA,拷贝数变更;M值,甲基化值。
不同亚型的患者面临不同的临床结局
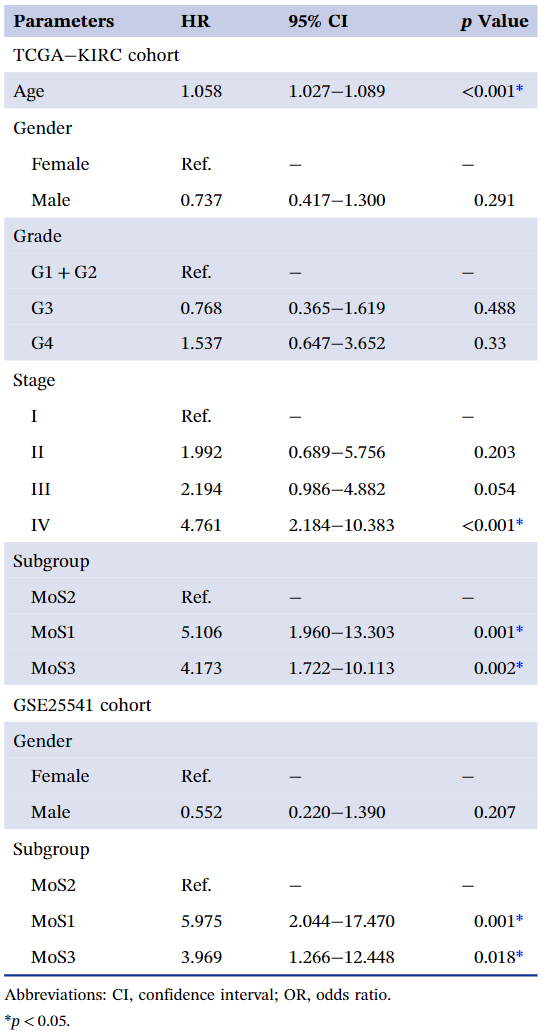
肿瘤患者的临床结果对于选择积极地根治性治疗还是消极地治疗非常重要,也可以让患者对未来的生活有一个预期。我们发现,MoS1亚型中处于G4肿瘤晚期的患者多于任何其他亚型(MoS1, 41.4% vs. MoS2, 4.7% vs. MoS3, 11.1%, p < 0.001, 表1),以及Stage IV(MoS1, 29.3% vs. MoS2, 7.0% vs. MoS3, 17.3¬¬%, p < 0.001, 表1)。同时,三个MoS中患者的平均年龄没有差异(MoS1,61.64 ± 11.93 vs. MoS2,60.99 ± 11.14,vs. MoS3,60.49 ± 11.44,p = 0.852,表1)。我们还观察了三种MoS亚型的预后对比,MoS2的OS较好,MoS1的预后较差,平均OS时间从MoS2、MoS3到MoS1逐渐降低(MoS1,37.74 ± 30.42 vs MoS2,56.39 ± 37.15,vs. MoS3,44.84 ± 33.45个月,p < 0.001,图1B),无进展生存间隔时间也有同样的趋势(MoS1,31.49 ± 31.79 vs. MoS2,47.25 ± 34.44,vs. MoS3,38.74 ± 31.67 个月,p < 0.001,图 1C)。
表1. 三种亚型的临床信息分布
MoS2中缺氧信号的沉默与良好的预后相关
为了进一步了解三种亚型的差异,我们进行了基因本体(GO)术语通路富集分析,以确定亚型特异性激活的信号通路。我们观察到MoS1患者存在膜蛋白靶向通路、胶原代谢和急性炎症反应通路的激活(图S2A、B)。MoS2中,内皮发育、脂肪酸代谢和分解代谢、mRNA加工的通路高度激活(图S2A、C)。同时,MoS3显著富集了免疫相关通路的激活,包括巨噬细胞迁移、抗原加工和呈递、细胞因子产生的调节以及免疫反应调节和细胞表面受体信号传导(图S2A、D)。据报道,缺氧与ccRCC的肿瘤发生密切相关。因此,我们评估了三种亚型ccRCC患者的缺氧状态,从Winter缺氧评分、Buffa缺氧评分和Ragnum缺氧评分,我们都观察到MoS2缺氧评分最低,代表较低的缺氧信号传导,与MoS2的良好预后一致(图 S2E)。
MoS3中存在激活的免疫微环境并与免疫治疗反应相关
我们通过CIBERSORT比较了MoSs中的免疫细胞浸润情况,MoS1中浸润的记忆 B 细胞、浆细胞、CD8+ T细胞、CD4+ T细胞和M0巨噬细胞最多。MoS2含有M1巨噬细胞、γδT细胞和嗜酸性粒细胞等促炎免疫细胞,这可能是MoS2良好预后的另一个原因。对于MoS3亚型,我们观察到单核细胞和抗炎M2巨噬细胞的高度浸润(图2A)。此外,我们将免疫微环境激活状态与之前发表的特征进行比较,发现在MoS1和MoS3中都有高度的免疫细胞浸润,但MoS1还含有显著激活的肿瘤浸润性T调节细胞(tumor-infiltrating T regulatory cell, TITR)和癌症相关的细胞外基质 (cancer-associated extracellular matrix, C-ECM) 特征,代表耗尽的免疫状态(图 2B)。至于免疫检查点的mRNA水平,我们发现 MoS3 比 MoS1 和 MoS2 含有更高的CTLA4水平(p = 0.004,图 2C),而MoS1比其他两种亚型表现出更高的PD-1水平(p = 0.0018,图 2D)。通过Submap算法,我们发现MoS3 中的患者可以从抗 PD-1 的免疫治疗中受益,但MoS1 和 MoS2则较少获益(图 2E)。通过最近模版预测(Nearest Template Prediction,NTP)分析选择了前300个特定标记基因,我们将分组情况映射到外部队列。为了验证 MoS3 更适合免疫治疗的新发现,我们收集了 CheckMate队列和Miao 队列的 mRNA 表达谱和抗 PD-1 治疗的反应结果。CheckMate队列中MoS3中71.4%的患者获得了临床获益,高于MoS1 (59.1%)和MoS2 (43.8%)的比例(图 2F)。在Miao队列,我们观察到类似的结果,80% 属于MoS3的患者有临床获益,但MoS1中的比例较低至38%,MoS2中为50%(图 2G)。
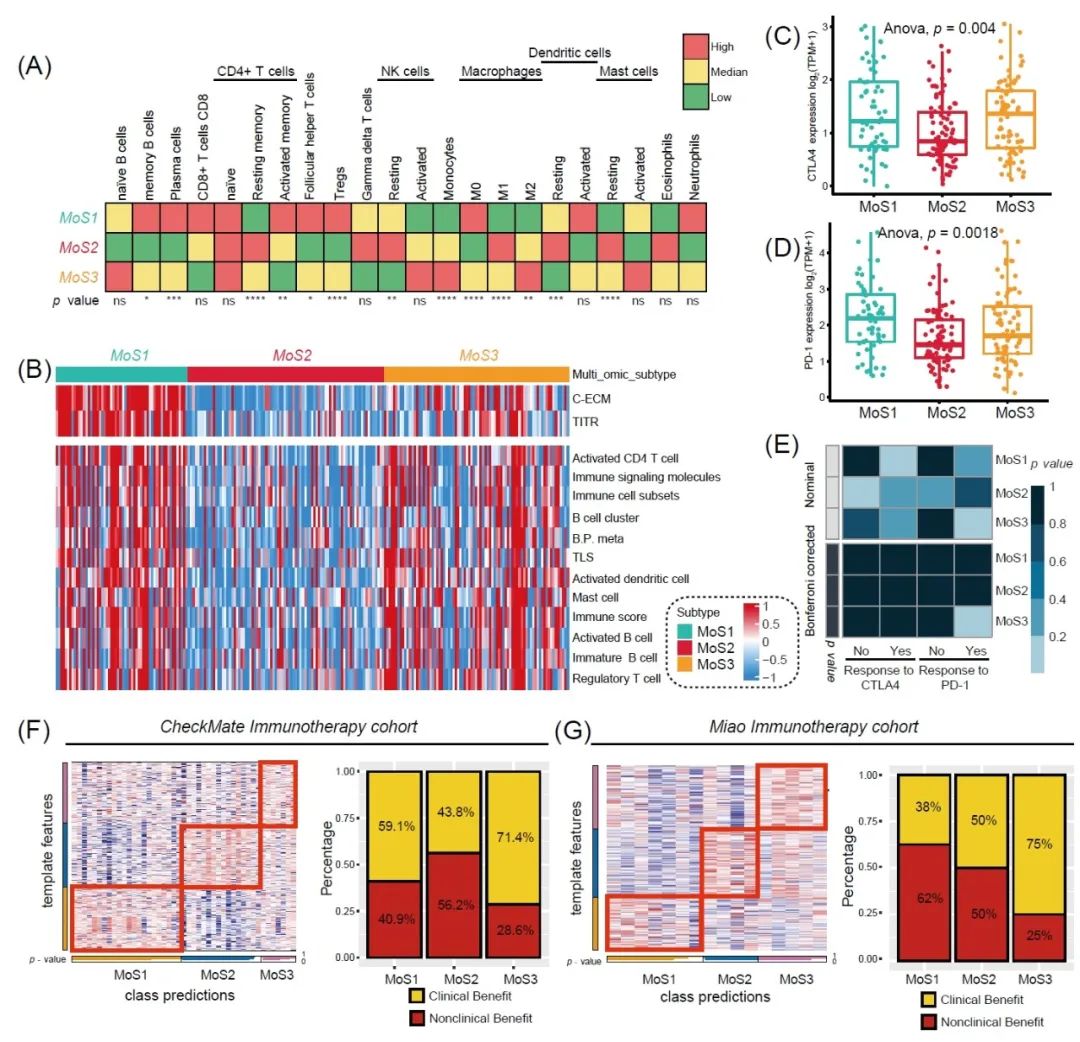
图2. 三种亚型之间的免疫微环境和免疫治疗反应差异
(A) MoSs中的免疫细胞浸润景观。(B)免疫微环境激活和耗尽状态以及 MoS1、MoS2 和 MoS3 亚型之间的特征的比较。(C) CTLA4表达水平的比较。(D) PD-1表达水平比较。(E) 子类映射图显示了三种MoS内对抗PD-1或抗CTLA-4治疗的不同反应。(F) 基于CheckMate免疫治疗队列评估 MoS1、MoS2和MoS3亚型对Nivolumab的反应。(G) Miao队列中MoS1、MoS2 和 MoS3 亚型对 Nivolumab 的反应评估。TITR,肿瘤浸润性T调节细胞;C-ECM,癌症相关细胞外基质;TLS,三级淋巴结构。
SETD2突变通过PLXNA2促进ccRCC患者不良预后
对于TCGA-KIRC 队列中225名患者的总体突变研究中,MoS1中86%的患者、MoS2中 91%的患者和MoS3中84%的患者至少含有一种基因突变(图 3A)。MoS2包含较高的PBRM1和VHL突变,而SETD2突变主要在MoS1和MoS3中观察到,这预示着预后不良(图3B)。对于SETD2突变和不利临床结果的潜在原因,我们首先观察到了SETD2突变导致SETD2 mRNA表达水平降低(p <0.001,图3C),并且较低的SETD2水平与较短的OS相关(p < 0.001,HR = 0.56,图 3D)。此外,我们选择了来自GSE135105的数据,其中包含从ccRCC细胞系生成的三个对照样本和三个siDETD2样本。结果表明siSETD2激活了多种免疫相关信号通路,包括抗原加工和呈递、细胞因子-受体相互作用和自然杀伤细胞介导的细胞毒性通路(图3E)。因此,SETD2突变较少和表达较高可能是MoS2中免疫细胞浸润不足的原因。我们构建了SETD2的ceRNA网络(图 3F),并揭示miR-497-5p、miR-655-3p和miR-374-5p是调节PLXNA2轴的枢纽组件(图 3G),敲低 SETD2上调表达三个 miRNA并进一步抑制PLXNA2水平,SETD2表达与PLXNA2表达呈正相关(R = 0.54,p < 0.001,图3H)。PLXNA2可能是SETD2突变的ccRCC患者的潜在治疗靶点。
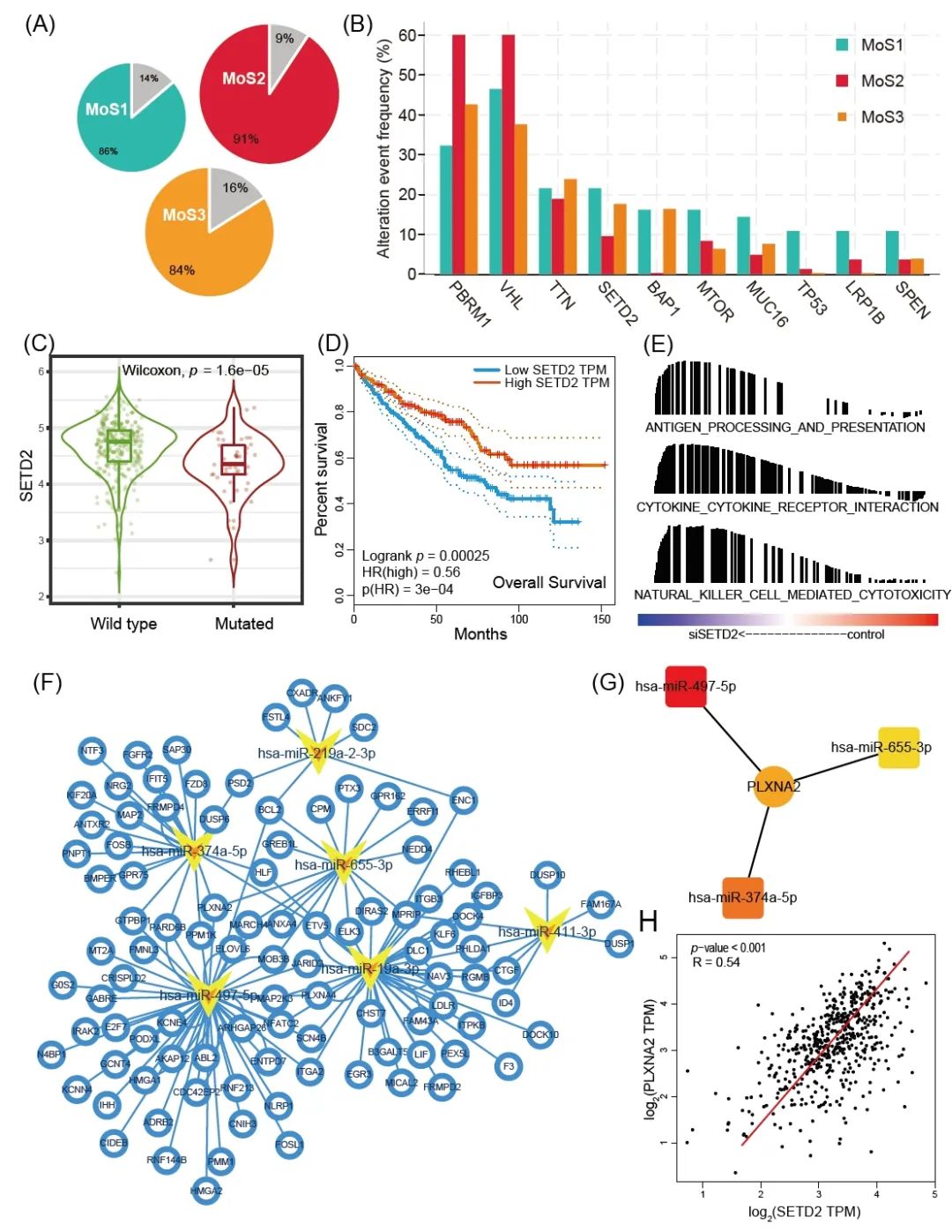
图 3. 三种亚型突变基因的阐明以及SETD2在ccRCC中的关键作用
(A) MoS1、MoS2和 MoS3亚型基因突变患者的比例分布。(B) 三种亚型之间前10个改变基因的频率的比较。(C) 野生型和突变型SETD2肿瘤之间SETD2表达的比较。(D) K-M图显示SETD2表达水平低的患者总体生存率较差。(E) 根据GSE135105数据集评估敲除SETD2后激活的免疫相关通路。(F) SETD2下游 miRNA和基因ceRNA网络分析。(G) SETD2通过miR-497-5p、miR-655-3p和miR-374-5p 调节PLXNA2的核心调节网络。(H) SETD2与PLXNA2的表达呈正相关。
SETD2的敲低促进ccRCC细胞的细胞增殖、迁移和侵袭
我们首先收集癌组织并通过免疫组织化学评估SETD2和PLXNA2的表达水平。通过计算SETD2和PLXNA2免疫组化评分,我们发现SETD2和PLXNA2表达呈显着正相关(n = 60,R = 0.37,p < 0.01,图4A-B)。此外,SETD2的敲除可以相应地抑制PLXNA2的表达水平(图4C)。这些结果证明PLXNA2可能是SETD2的下游靶标。此外,CCK8和伤口愈合实验表明,SETD2的敲除促进了RCC细胞的增殖和迁移能力(图4D)。与上述结果一致,SETD2的抑制也延缓了这些细胞在侵袭和跨孔实验中的迁移(图 4E)和侵袭(图 4F)。总之,这些结果表明SETD2可能在ccRCC中发挥肿瘤抑制因子的作用。
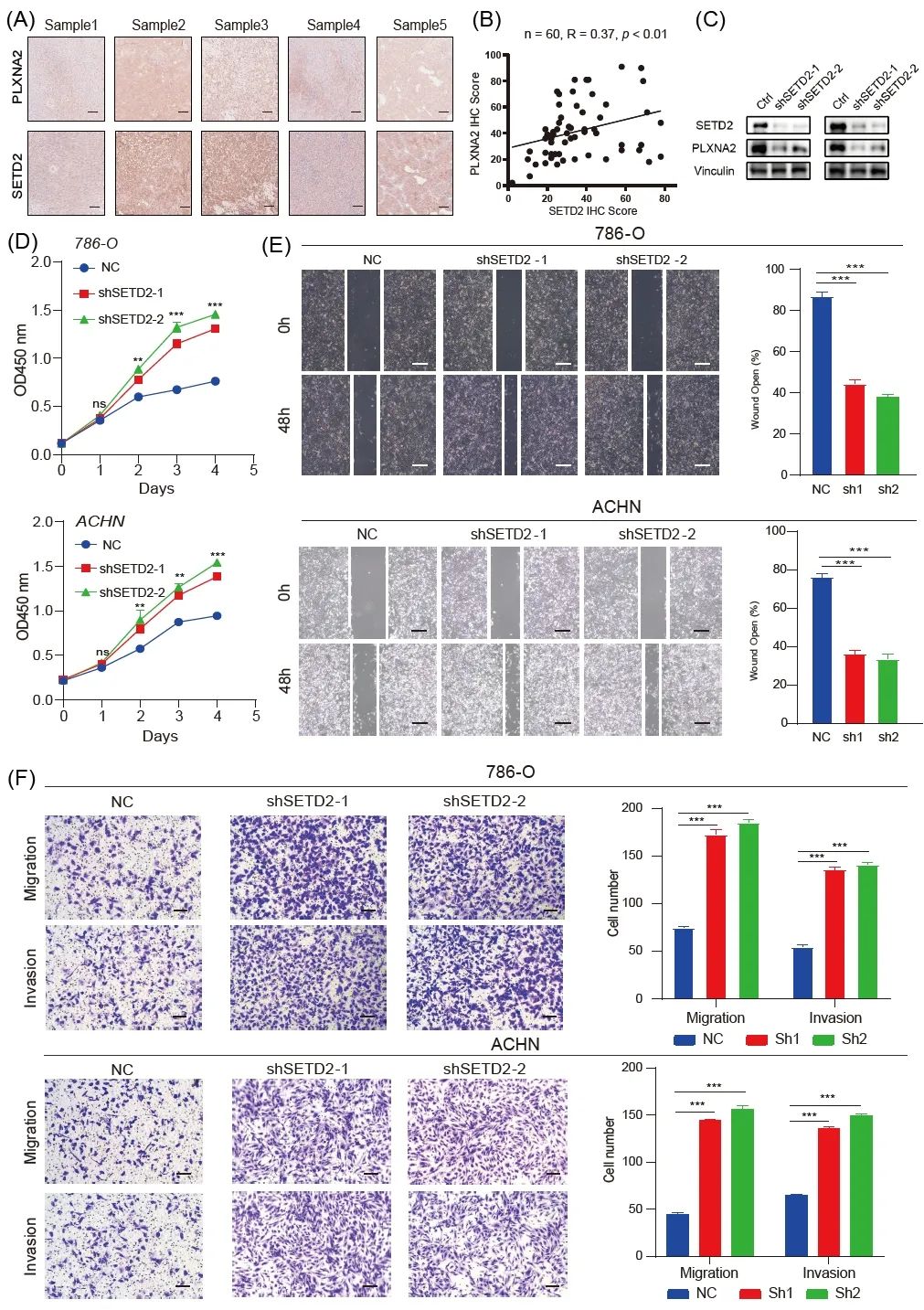
图4. 验证SETD2在肿瘤组织、786-O和ACHN ccRCC细胞系中的功能
(A) 通过免疫组织化学分析 60 例 ccRCC 患者癌组织中 SETD2 和 PLXNA2 的表达。比例尺,300 μm。(B) SETD2 和 PLXNA2 IHC 评分的相关性。(C) SETD2 的敲低抑制了 PLXNA2 的表达。(D) 转染阴性对照和 sh-SETD2 后 786-O 和 ACHN 的细胞增殖。(E) 表达载体对照或 sh-SETD2 的 786-O 和 ACHN 细胞的伤口愈合测定。比例尺,200 μm。(F) SETD2 对照和敲低 786-0 和 ACHN 细胞中细胞侵袭和迁移数量的比较。比例尺,100 μm。ns,不显着;*,p < 0.05;**,p < 0.01;***,p < 0.001;NC,阴性对照。
舒尼替尼更适合MoS2,AKT抑制剂更适合MoS1
为了确定ccRCC患者的精准化疗药物,我们首先评估了对舒尼替尼的反应,依赖于 GDSC平台,我们发现MoS2亚型对舒尼替尼治疗有更高的敏感性(p = 0.089,图 5A)。此外,我们在E-MTAB-3267队列中识别出MoS,该临床试验队列包含53名接受舒尼替尼治疗的ccRCC患者(表 S2)。舒尼替尼治疗后,MoS2患者的OS结局最好,MoS1患者的预后最差(p = 0.0037,图 5B)。对于ccRCC治疗的其他常用化疗药物,我们也进行了评估,发现阿西替尼、GDC0941和二甲基草酰甘氨酸更适合MoS1患者的治疗(均p < 0.05,图5C)。我们进一步从GDSC在线网站获得了针对SETD2突变细胞的敏感药物,我们发现SETD2突变细胞对PI3K/AKT信号通路抑制剂敏感(AZD8186,AZD5363,Alpelisib,图5D),这进一步得到PI3K/AKT抑制剂的验证 MK.2206 (p < 0.001) 和 AZD6482 (p < 0.001)(图 5E)。
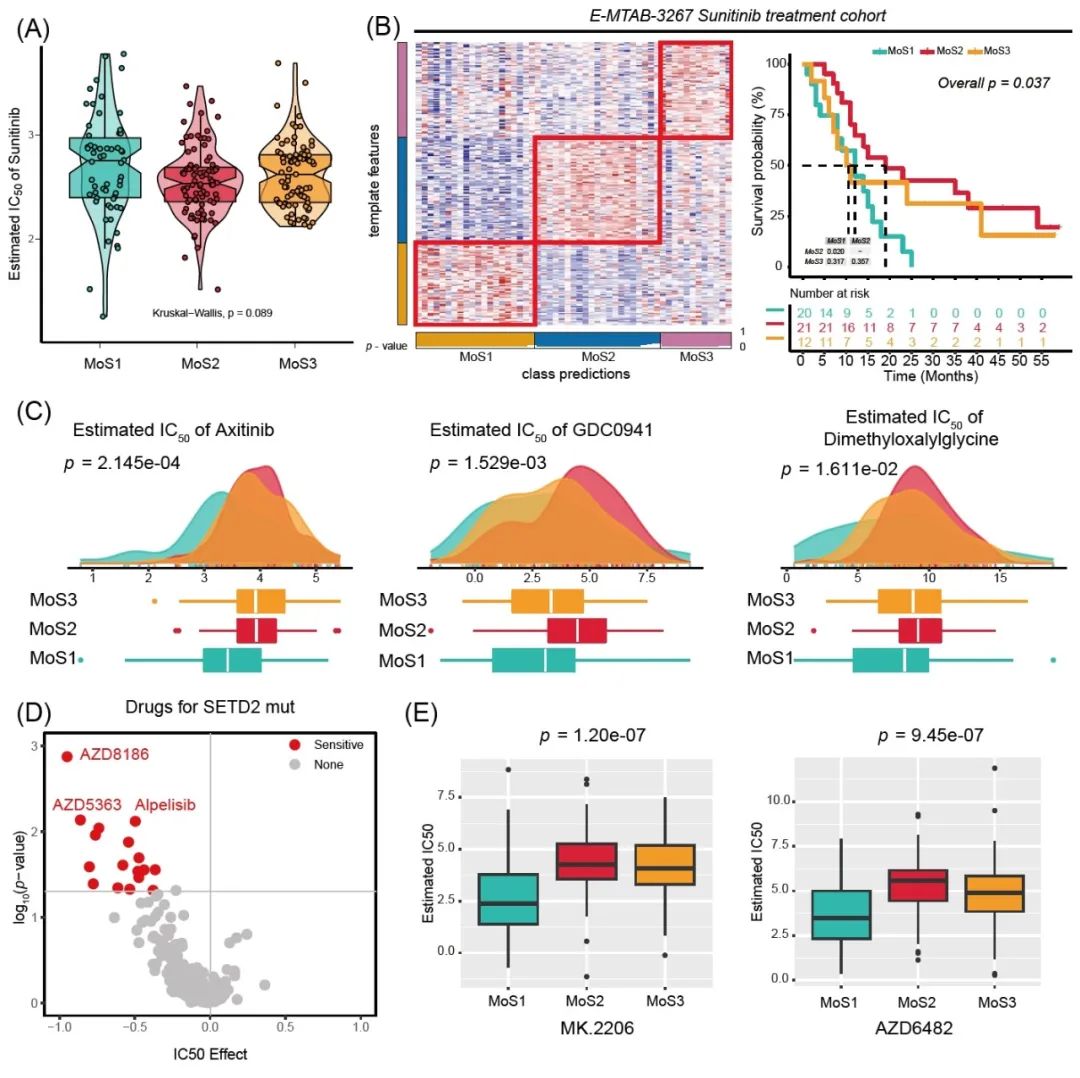
图 5 确定适合 MoS1 和 MoS2 亚型的分子药物
(A) TCGA-KIRC MoS亚型中舒尼替尼治疗IC50值的预测;(B) MoS 亚型聚类和 Kaplan-Meier 图显示 E-MTAB-3267 舒尼替尼治疗队列中不同的总体生存率;(C) TCGA-KIRC MoS亚型中阿西替尼、GDC0941和二甲基草酰甘氨酸治疗IC50值的预测;(D) SETD2突变亚型潜在化疗药物的敏感性预测;(E) TCGA-KIRC MoS 亚型中 PI3K/Akt 抑制剂治疗IC50值的预测。
外部队列中重现MoS并验证不同的临床结果
利用300个亚型特异性标记,我们在外部队列中重现了MoS分类。GSE22541的40名患者被分为MoS1、MoS2、MoS3,其中MoS2的患者预后最好,而MoS1患者的临床结果最差(图6A)。进一步的多因素cox回归分析调整了性别偏倚,MoSs仍然是ccRCC的独立预后因素(表2)。在GSE40435中,MoS2中的患者大多处于早期肿瘤阶段(I期,10.42%;II期54.17%),这可能表明预后良好,而MoS1包含大多数IV期患者(19.23%)(图6B)。GSE53757的结果显示出类似的趋势,约50%的患者属于早期I期的MoS2,而33.33%的患者属于IV期的MoS1(图 6C)。
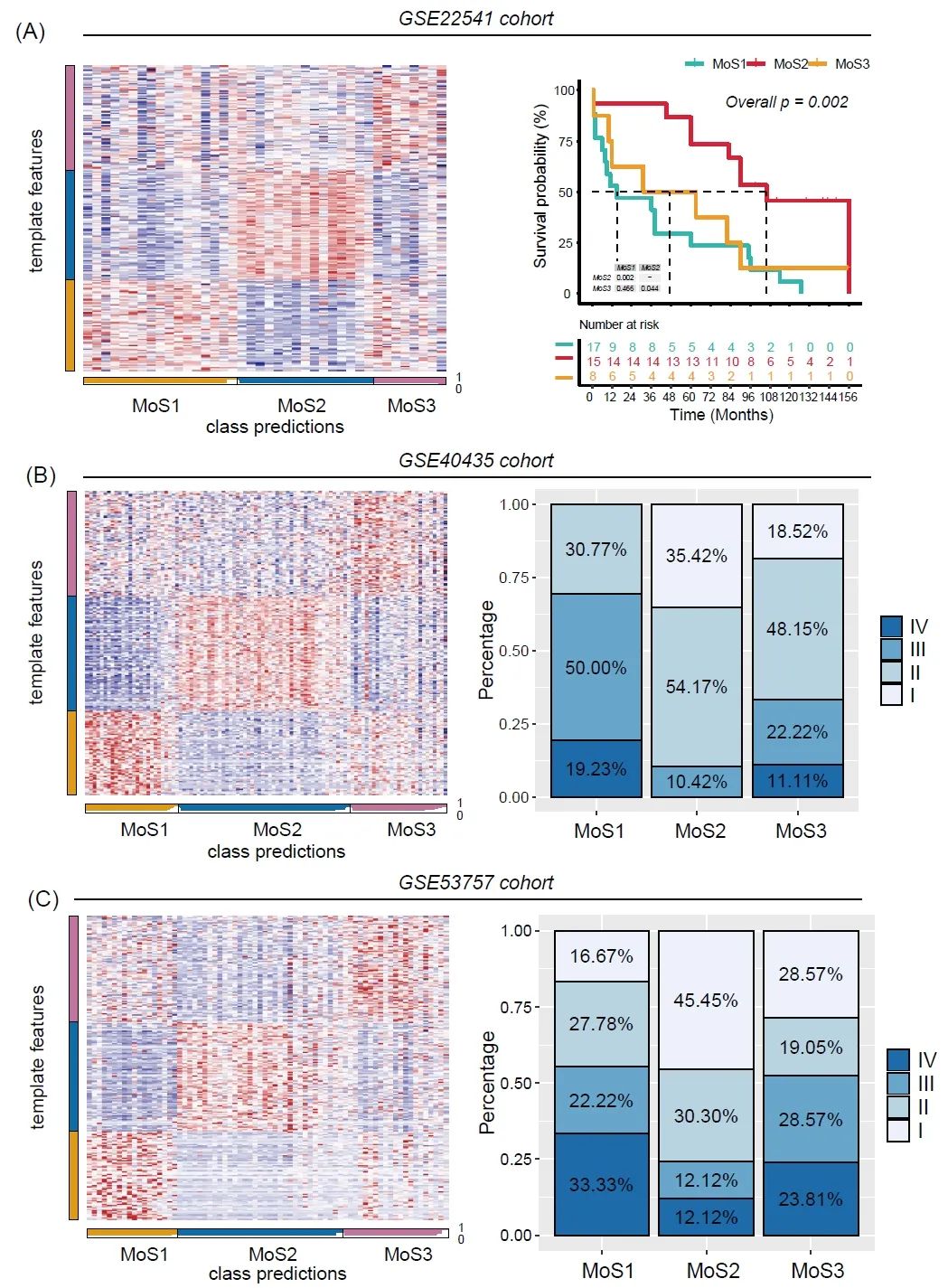
图 6. 通过亚型特异性标记对MoS分类进行外部验证
(A) GSE22541 队列中的 MoS 分类,并通过 Kaplan-Meier 图评估不同的总体生存率;(B) GSE22541 队列中的 MoS 分类及肿瘤分期的分布;(C) GSE53757 队列中的MoS分类及肿瘤分期的分布。
表2. 新定义的癌症亚型是 ccRCC 患者的独立预后因素
讨 论
尽管通常认为ccRCC是一种可早期检测的疾病,且可以通过消融治疗或切除策略治愈,但三分之一的患者可能会出现转移,并发展到终末阶段。与其他肿瘤类似,ccRCC 预后的异质性源于内在的分子改变。目前,高通量测序和生物信息学的发展促进了对ccRCC中分子景观的全面阐明。目前已经基于不同分子改变建立了许多新颖的风险分层方案,例如,TRACER肾脏系统序将ccRCC分为七种主要亚型,包括VHL单驱动、PBRM1-SETD2、PBRM1、PBRM1-PI3K、VHL野生型、多克隆驱动和BAP1驱动亚型,这些基因在不同的亚型中表现出明显的差异预后;Peng及其同事基于甲基化特点构建分类系统来预测 ccRCC的生存率,高甲基化评分组比低甲基化评分组表现出更长的OS(HR = 2.46,95% CI:1.63 – 3.71,p < 0.001)。另一方面,ccRCC中不同组学的改变可能会阻碍可靠的风险分层方案的识别,因此,我们必须基于多种视角揭示分子亚型,以克服目前仅单一组学造成的局限。
应用十种鲁棒性的聚类算法来研究综合数据与OS之间的相关性,我们建立了一个共识分类系统,该分类充分考虑了ccRCC细胞多个组学的异质性,包括 mRNA、lncRNA 表达、CNA、DNA甲基化和基因的特征突变。在定义的三个亚型中,MoS1亚型预后最差,MoS2亚型预后最好,MoS3亚型预后中等。此外,这三种不同的表型表现出显著不同的分子改变景观和信号通路激活,从而导致不同的代谢过程和生物学行为。
恶性肿瘤的特点是循环缺氧,因为它们的快速生长与血管的不匹配发育相对应,而缺环境会增加肿瘤的生长速度,从而促进肿瘤的迁移和转移。与MoS1和MoS3相比,MoS2表型具有较低的缺氧评分,强烈表明MoS2代表相对惰性和低侵袭性亚型。VHL突变是ccRCC 的标志性癌基因。VHL突变与ccRCC预后之间的相关性存在争议,有正相关、负相关和无关相关等报道。一项包含五项VHL突变相关研究的荟萃分析显示,突变VHL与ccRCC预后之间没有相关性,但具有保护性趋势。在本研究中,更多的 VHL 突变富集到 MoS2 亚型中,与其他表型相比,该亚型与更长的OS相关,这也表明了保护性趋势。MoS1和MoS3 表型表现出高DNA甲基化负担。DNA甲基化在不改变DNA序列的情况下修改遗传信息并影响表达,增强子和启动子CpGs的高甲基化有助于大量抑癌基因的表观沉默,从而促进ccRCC的增殖、侵袭和转移。CNA涉及染色体丢失或增益,可影响多个基因,并且随着剂量的积累,相关基因的表达水平将随之改变。我们的结果表明,9号染色体缺失是ccRCC的另一个恶性事件,与晚期阶段和不良临床结果有关。这一发现与之前的几项研究一致,可能是由于9号染色体存在几种抑癌基因,包括9p21上的CDKN2A,位于9p13上的CAIX,位于9p24.1-p13.3上的NDUFB6。
SETD2位于3p,编码H3赖氨酸36组蛋白甲基转移酶的表达,并在ccRCC中首次报道。SETD2缺陷会通过多种机制促进肿瘤发展,例如抑制DNA修复、改变细胞机制、激活癌基因和扰乱细胞周期进程。作为ccRCC的保护因素,34.07% 的患者存在SETD2突变,并且与侵袭表型相关。在本研究中,在MoS1和MoS3亚型中观察到SETD2的高突变,这与它们较差的临床表现相一致。这些证据表明SETD2突变是促进ccRCC进展的关键事件,因此我们进行了进一步的研究。基于ceRNA网络分析,我们揭示了下游关键基因PLXNA2,它可以被SETD2通过miR-497-5p、miR-655-3p和miR-374-5p调节。IHC验证了SETD2和PLXNA2之间的正相关性,并且在STED2敲低的786-O和ACHN细胞系中,ccRCC呈现出PLXNA2显著降低,证明了ceRNA网络的结果。同时,STED2敲低的ccRCC细胞表现出高增殖、迁移和侵袭的趋势,再次证实了SETD2在ccRCC中作为肿瘤抑制因子的功能。
我们进一步研究了三种MoS亚型的免疫细胞浸润状态和PD-1表达水平。MoS1具有最高的PD-1和PD-1表达水平,但伴随着 TITR 和 C-ECM 特征的激活,其表现出免疫原性“冷”表型,因此从理论上讲应该对PD-1阻断反应较低。同时,尽管免疫抑制途径的激活相对较少,但与MoS1亚型的患者相比,本研究中MoS3亚型的患者实际上会从抗 PD-1治疗中获益更多。与我们的预期一致,PD-1的表达是决定抗PD-1治疗疗效的方面之一,但免疫激活状态也起着枢纽作用。C-ECM、TITR是免疫耗竭状态的标志,MoS1高耗竭评分表明抗PD-1治疗反应较差。相比之下,MoS3细胞中免疫细胞的高浸润意味着免疫状态激活以及对抗 PD-1治疗的高反应。不同类型的巨噬细胞浸润可能是MoS亚型之间预后不同的原因之一,M1巨噬细胞在MoS2表型中高度富集,正向调节免疫反应,从而杀死肿瘤细胞,而MoS3中具有大量的M2巨噬细胞浸润,分泌多种生长因子并重塑肿瘤微环境以支持肿瘤进展、侵袭和转移。
局部晚期或转移性疾病占所有ccRCC病例的三分之一左右,并且对细胞因子治疗、化疗或放疗等典型辅助治疗的敏感性有限。ccRC的高度血管化特征为抗血管生成治疗提供了机会,舒尼替尼已被用作转移性ccRCC的标准一线治疗方案。然而,超过一半的患者无法从舒尼替尼中获益,因此,识别潜在敏感性患者将提高舒尼替尼的疗效。在这项研究中,MoS2患者对舒尼替尼表现出高度敏感,而MoS1则表现出较差的反应性。对于MoS1亚型患者,阿西替尼、GDC0941和二甲基草酰甘氨酸更有可能延缓ccRCC的进展。PI3K-AKT 通路被广泛报道为SETD2缺陷的ccRCC中一个有前途的药物靶点,表明 PI3K-AKT 通路的抑制剂可能适合MoS1和MoS2亚型,因为它们具有高SETD2突变,并且基于GDSC在线网站,我们还鉴定了几种针对MoS1的药物,例如AZD8186、AZD5653和Alpelisib。总的来说,上述结果表明 MoSs 分类器可以为不同患者制定个体化治疗策略。
结 论
综上所述,我们通过多组学特征建立了新型ccRCC分子分类。三种MoS亚型表现出显著不同的临床病理参数、分子改变、信号激活、免疫细胞浸润和治疗反应相关。MoS1亚型预后最差,可能是由于免疫微环境衰竭、缺氧特征激活所致,但可以受益于PI3K/AKT抑制剂治疗。MoS2亚型的VHL和PBRM1突变较多,预后良好,更适合舒尼替尼治疗。MoS3是免疫“热”亚型,可以从抗PD-1免疫治疗中受益。我们还证实SETD2是ccRCC中的抑癌基因,随着肿瘤晚期SETD2蛋白水平降低,敲低SETD2导致细胞增殖、迁移和侵袭的促进。为了进一步测试MoS分类的价值,我们迫切需要进行纵向和前瞻性研究来提供更多证据。
方 法
多组学数据收集
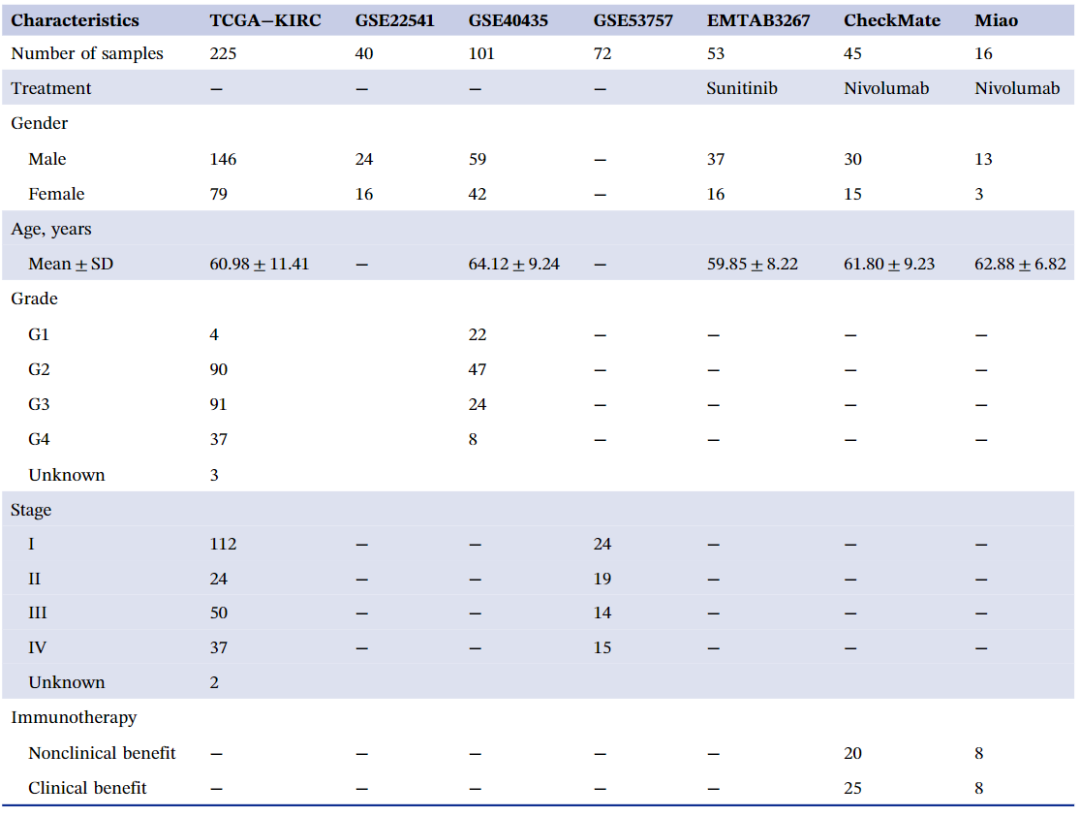
TCGA-KIRC队列被纳入多组学分析,其包含完整的转录组表达谱、生存结果、拷贝数改变、DNA甲基化和体细胞突变数据。我们使用R包“TCGAbiolinks”下载mRNA和lncRNA 表达谱。LncRNA数据被记录为非编码、3prime重叠ncRNA、反义RNA、lincRNA、有义内含子、有义重叠、lncRNA和双向启动子lncRNA组,并根据Vega (http://vega.archive.ensembl.org/) 进行鉴定。这些转录组的Ensembl ID 通过 GENCODE27转换为基因符号。我们获得了Illumina DNA 甲基化数据 (https://xenabrowser.net/)。由于缺乏大量TCGA-KIRC 患者的数据,miRNA 数据被删除。此外,体细胞突变、临床病理学特征均从cBioPortal (https://www.cbioportal.org/) 检索。为了获得训练队列,我们对患者进行了筛选,仅纳入了具有五个组学数据和总生存结果的ccRCC患者,最终总共留下了225名患者用于后续分析。为了验证训练队列的结果,我们收集了七个微阵列数据集。GSE22541、GSE40435和GSE53573队列被用于验证集。来自CheckMate 队列的45名患者和来自 Miao 队列的16名接受 Nivolumab(抗 PD-1)治疗的患者被纳入以评估不同ccRCC对免疫治疗的敏感性。E-MTAB-3267队列中纳入了53名接受舒尼替尼治疗的患者,以揭示舒尼替尼治疗的有效性。根据相应平台的注释文件,将每个入组队列的探针ID转移到基因符号中。如果基因符号用多个探针ID注释,则用计算平均值作为表达数据。表3和表S3列出了入组队列的所有临床病理学特征。
表3.入组队列的基本临床特征
原始数据整合和分子亚型分类
我们基于 mRNA 表达、lncRNA 表达、miRNA 表达、DNA 甲基化、CNA和体细胞突变数据的多组学数据建立了ccRCC的新分类系统。我们对转录组数据进行了log2(TPM + 1)计算,以使其更具可比性,对于DNA甲基化数据,我们只关注位于启动子在CpG岛的探针,并且对于具有多个探针映射到其启动子的基因,中均值代替。对于基因突变数据,保留基因突变矩阵中非同义变异的样本用于后续分析,包括移码删除/插入、框内删除/插入、错义/无义/不间断突变、剪接位点或翻译起始位点突变。对于 CNA,我们按照文献压缩了基因组片段,删除了平坦特征以加快聚类效率。为了更好地将亚型与其相应的临床信息匹配,我们根据Cox回归生存分析提取与OS最相关的因素,考虑P值小于0.001的因素,最终入组664例 mRNA(表S4)、60个lncRNA(表S5)、60个CNA(表S6)、569个DNA甲基化位点(表S7)。突变频率高于5%的14个基因也被纳入多组学分析。为了找到最佳的分组数量,计算了聚类预测指数(CPI)和间隙统计,并参考了先前报道的 RCC 分子亚型。随后,进行了10个最先进的多组学来建立最终的分类模型,包括 iClusterBayes、moCluster、CIMLR、IntNMF、ConsensusClustering、COCA、NEMO、PINSPlus、SNF和LRA,如R包中所述“ MOVICS”是我们最近开发的。并且进一步整合上述算法的聚类结果来生成鲁棒性聚类。
对于七个外部队列,借助NTP,将表达数据分成由训练队列生成不同簇。选择按log2FoldChange排序的差异最大的基因作为每个亚型的生物标志物,这些生物标志物应通过显著性阈值(调整后的P< 0.05)检验,并且不得与为其他亚型识别的任何生物标志物重叠。
评估不同分子特征
对于所有新定义的分子亚型,为了揭示潜在的生物学特征,我们借助基因集变异分析(Gene set variation analysis, GSVA)计算GO通路的富集分数来表征亚型,确定的亚型特异性通路应满足显著性阈值,调整后的P< 0.25,并且任何与其他亚型重叠的通路都将被删除。我们展示了前10个GO通路和亚型之间的关系网络。
至于免疫细胞丰度,我们使用 CIBERSORT算法对每位患者进行评估,并通过热图可视化每个亚型患者的平均浸润率,以表示不同的免疫景观。我们还评估了之前研究中使用的免疫相关特征,以进一步区分免疫激活和免疫耗尽特征。每个签名的富集分数由 R 包“GSVA”计算,并进一步用 R 包“ComplexHeatmap”显示。
对于不同亚型的CNA 和基因突变,MIBC-TCGA 队列的拷贝数改变基因组(Copy number-altered genome, FGA)的个体分数根据拷贝数片段数据计算如下:
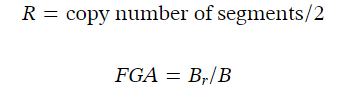
FGA是log2(拷贝数)值大于0.3的基因组与拷贝数分析的基因组的比例,其中Br表示|log2 R|>0.3的片段中的碱基数,B表示所有片段中的碱基数。cBioProtal显示不同亚型之间的改变事件,包括CNA和基因突变,以及获得或丢失的CNA是特定染色体。winter缺氧评分、buffa 缺氧评分和ragnum缺氧评分也从cBioProtal下载。通过Wilcoxon检验比较野生型和突变型SETD2样品之间的改变事件频率。
预测精准治疗策略
我们评估了集群患者对化疗的疗效,包括常规ccRCC化疗、抗缺氧治疗和PI3K/Ak通路抑制剂。为了估计不同个体的化疗反应,使用来自GDSC (https://www.cancerrxgene.org/)的727个人类癌细胞系(Human cancer cell lines, CCL)作为训练队列,并使用R包“pRRohetic”来预测相应的灵敏度。通过岭回归计算半数最大抑制浓度(Half maximal inhibitory concentration, IC50),并将其设置为疗效评定。两种常见化疗药物(索拉非尼和舒尼替尼)、三种抗缺氧药物(阿西替尼、GDC0941 和二甲基草酰甘氨酸)和两种 PI3K/Akt 通路抑制剂(MK.2206 和 AZD6482)的治疗反应分别通过 IC50 进行量化。IC50越低表明治疗敏感性越高,采用10倍交叉验证来衡量预测效果。如上所述,E-MTAB-3267队列中纳入了53名接受舒尼替尼治疗的患者,以揭示舒尼替尼治疗的有效性。我们还关注抗PD-1的免疫治疗。纳入了47名接受抗CTLA-4或抗PD-1检查点抑制治疗的黑色素瘤患者,其中包含 795 个免疫相关基因谱。子类映射分析(GenePattern 模块“SubMap”)揭示了独立数据集中的常见亚型,用于检测亚型之间基因表达谱的相似性。此外,我们还纳入了CheckMate队列中的45名患者和Miao队列中接受Nivolumab(抗 PD-1)治疗的16名患者,以评估对免疫治疗的敏感性。
统计分析
所有统计分析均使用 R 版本4.0.2 (https://www.r-project.org/) 进行。对于连续变量,如果数据呈正态分布,则使用学生t 检验和双样本 Mann-Whitney 来比较两组,或者应用威尔逊等级检验。在比较分布的正态性之前,将进行 Shapiro-Wilk 检验。对于分类变量,将进行卡方检验和费舍尔精确检验。为了比较簇间基因突变频率,进行了排列检验。依靠对数秩检验,Kaplan-Meier 来分析总生存OS。建立Cox回归模型计算风险比(Hazard Ratio, HR)值和95%置信区间(confidence interval CI)。上述大部分分析都是基于R包“MOVICS”进行的。为了评估簇之间的生物学差异,使用基于预排序基因列表的R包“clusterProfiler”执行 GSEA,该列表由“limma”包中的 log2FoldChange 算法排序。进行Pearson相关性检验来计算系数值,p < 0.05被认为具有统计显着性。
代码和数据可用性
本研究中使用的公共数据可在 TCGA、ArrayExpress 和 GEO 中获取。本研究中的分析过程嵌入在 R 包“MOVICS”中,位于https://github.com/xlucpu/MOVICS,我们最近为多组学集成和可视化开发了它。本研究的代码和数据可在Github上获取(https://github.com/AHMUJia/ccRCC_MOS)。补充材料(图形、表格、脚本、图形摘要、幻灯片、视频、中文翻译版本和更新材料)可以在在线 DOI 或 iMeta Science 中找到http://www.imeta.science/。
引文格式:
Jialin Meng, Aimin Jiang, Xiaofan Lu, Di Gu, Qintao Ge, Suwen Bai, Yundong Zhou, Jun Zhou, Zongyao Hao, Fangrong Yan, Linhui Wang, Haitao Wang, Juan Du, Chaozhao Liang. 2023. Multi-omics characterization and verification of clear cell renal cell carcinoma molecular subtypes to guide precise chemotherapy and immunotherapy. iMeta e147. https://doi.org/10.1002/imt2.147
作者简介

孟佳林(第一作者)
● 安徽医科大学校聘副教授,硕士生导师,美国罗切斯特大学访问学者。
● 主要研究方向:泌尿系统肿瘤及慢性前列腺炎的诊断与治疗。主持国家自然科学基金1项;参与编译第11版《坎贝尔-沃尔什泌尿外科学》;在Journal of Infection,Advance Science,Clinical and Translational Medicine等国际期刊发表相关研究成果,并获得发明专利一项。

江爱民(第一作者)
● 海军军医大学第一附属医院泌尿外科2022级博士。
● 主要研究方向为泌尿系统肿瘤耐药及转移相关标志物及机制研究,目前以第一作者(含共一)在eClinicalMedicine,Cell & Bioscience,MedComm,Molecular Cancer等期刊发表论文10余篇。

陆晓凡(第一作者)
● 法国斯特拉斯堡遗传与分子生物研究所博士后,美国MD安德森癌症中心访问学者。
● 主要研究方向为罕见肿瘤/肾癌的多组学分型,以及借助统计模型在肿瘤临床试验队列确定影响治疗响应的表观遗传因素。开发MOVICS多组学分子分型及可视化R包。在Cell Reports Medicine, Nature Communications, Clinical Cancer Research, Genome Biology,Bioinformatics等国际学术期刊发表相关成果。

杜鹃(通讯作者)
● 香港中文大学(深圳)医学院助理教授,助理院长,博士生导师。
● 主要研究领域:肿瘤外泌体分泌机制与靶向递送,基于临床大数据的疾病诊疗标记物筛选等。在JASN,Hypertension,Mol Ther Nucleic Acids等杂志发表SCI论文60余篇,主持国家自然科学基金4项,获得省科技成果奖三等奖1项。

梁朝朝(通讯作者)
● 安徽医科大学副校长,主任医师、教授、博士生导师,兼任中国医师协会男科与性医学医师分会会长、安徽省医师协会会长。
● 担任Asian Journal of Andrology等20余家杂志(常务)编委及国家科技奖、国家自然科学基金等评审专家;入选新世纪百千万人才工程国家级人选,获全国优秀科技工作者荣誉称号;以主持人身份获省部级科技奖励一等奖4项、二等奖4项,累计主持国家自然科学基金重点项目1项、面上项目8项;累计培养博士后、博士、硕士研究生150余名。
更多推荐
(▼ 点击跳转)
iMeta | 引用7000+,海普洛斯陈实富发布新版fastp,更快更好地处理FASTQ数据
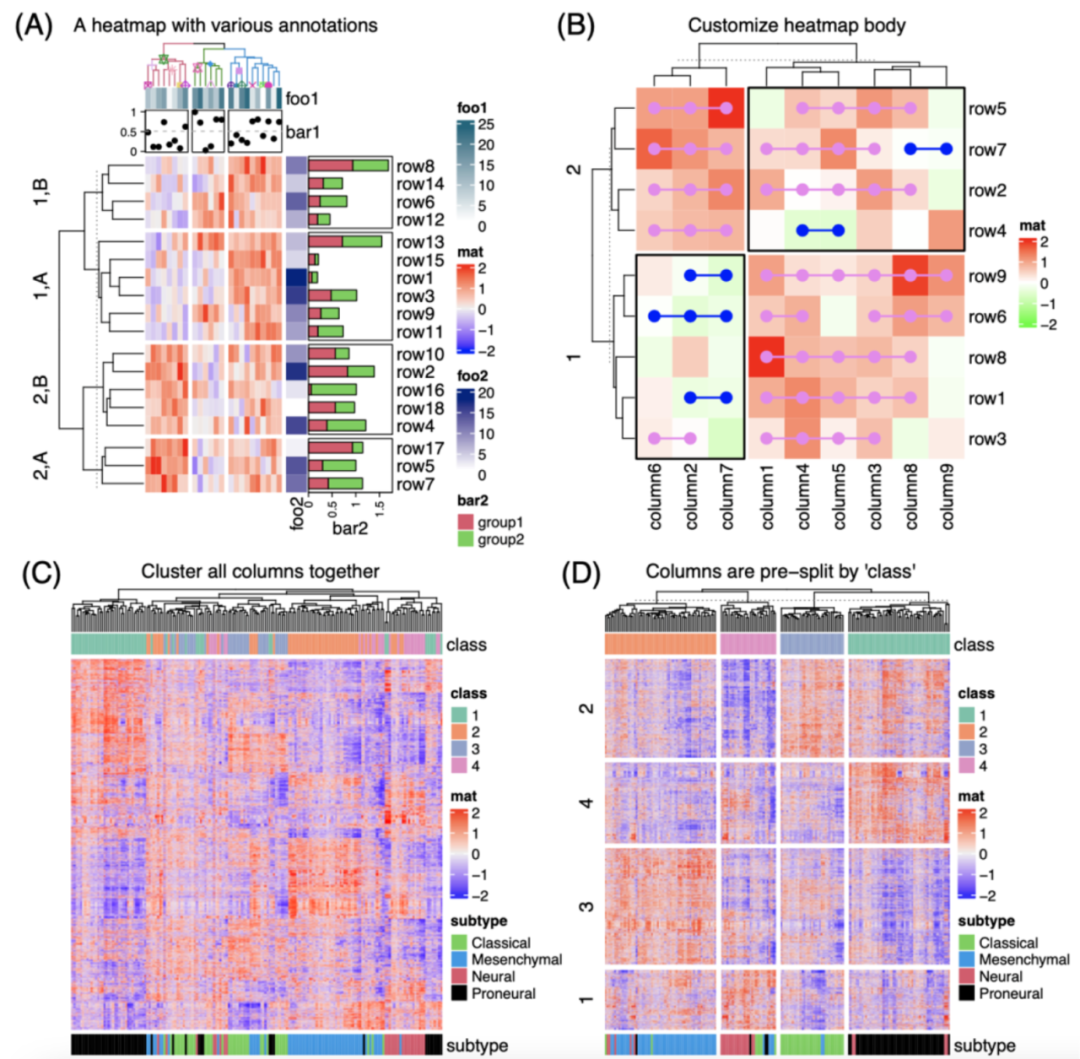
iMeta | 德国国家肿瘤中心顾祖光发表复杂热图(ComplexHeatmap)可视化方法

1卷1期

1卷2期

1卷3期

1卷4期

2卷1期

2卷2期

2卷3期

2卷4期
期刊简介
“iMeta” 是由威立、肠菌分会和本领域数百位华人科学家合作出版的开放获取期刊,主编由中科院微生物所刘双江研究员和荷兰格罗宁根大学傅静远教授担任。目的是发表原创研究、方法和综述以促进宏基因组学、微生物组和生物信息学发展。目标是发表前10%(IF > 15)的高影响力论文。期刊特色包括视频投稿、可重复分析、图片打磨、青年编委、前3年免出版费、50万用户的社交媒体宣传等。2022年2月正式创刊发行!
联系我们
iMeta主页:http://www.imeta.science
出版社:https://onlinelibrary.wiley.com/journal/2770596x
投稿:https://mc.manuscriptcentral.com/imeta
邮箱:office@imeta.science

















 1567
1567

 被折叠的 条评论
为什么被折叠?
被折叠的 条评论
为什么被折叠?
 到【灌水乐园】发言
到【灌水乐园】发言









