







- 最新下载链接包含参考基因组和对应注释文件
https://www.ncbi.nlm.nih.gov/datasets/genomes/?txid=9606
下载数据(人类)
NCBI
由于我需要获取剪切位点两边的序列,那么我需要下载参考基因组数据和注释文件。参考基因组下载常用的有ncbi、ucsc和ensemble。下图是参考基因组版本对应信息。
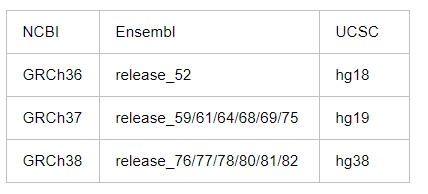
我是从NCBI下载
- 链接(https://www.ncbi.nlm.nih.gov/)

- 点击搜索之后,就可以在页面中找到了。

GenCode
我的注释文件是在GenCode下载,下面为版本信息。跟上面下载的参考基因组版本对应就行。

- 然后,如下图所示,我现在的该注释文件,GTF文件格式。

下载完之后,我们可以发现注释文件的规律。一个基因号下面是多个转录本,每个转录本下面是外显子,外显子又会分为UTR和CDS。如下图所示:

注释文件中是没有内含子信息,但是通过上图我们可以发现,基因中除了外显子就是内含子。所以我们可以根据注释文件中基因的位置和外显子的位置来确定内含子的位置。
bedtools
获取内含子位置
通过下述代码获取内含子位置,注意修改注释文件名称。
# 安装依赖
$ conda install bedtools
$ conda install bedops
# Gene annotation file gencode.v19.annotation.gtf
GENE_ANNOT_FILE='gencode.v19.annotation.gtf'
# Extract genes
awk '$3 == "gene"' $GENE_ANNOT_FILE > genes_temp.gtf
# Use BEDops convert2bed
# convert2bed won't work if the "transcript_id" field is not there. This makes an empty "transcript_id" field so convert2bed is happy.
awk '{ if ($0 ~ "transcript_id") print $0; else print $0" transcript_id \"\";"; }' genes_temp.gtf > genes.gtf
#Extract exons (exons always have transcript_id field, no need to repeat above)
awk '$3 == "exon"' $GENE_ANNOT_FILE > exons.gtf
#Convert both to bed format so we can use bedtools
convert2bed -i gtf < exons.gtf > exons.bed
convert2bed -i gtf < genes.gtf > genes.bed
# Remove exons from the genes.bed file, enforce strand specificy (-s)
bedtools subtract -a genes.bed -b exons.bed -s > introns.bed
# Convert intron bed file to GTF file
# 最终结果
awk '{print $1"\t"$7"\t""intron""\t"($2+1)"\t"$3"\t"$5"\t"$6"\t"$9"\t"(substr($0, index($0,$10)))}' introns.bed > introns.gtf
# combine with original annotations file
cat gencode.v19.annotation.gtf introns.gtf > gencode.v19.introns.gtf
# remove generated temporary files
rm genes.gtf exons.gtf exons.bed genes.bed introns.bed
获取内含子序列
利用bedtools 里面的
在获取内含子位置之后,本以为获取内含子序列会非常快速。但是,被窝瞎搞,浪费了一整天时间。一直注视文件和参考基因组匹配不上,报错:
WARNING. chromosome (chromosome Y) was not found in the FASTA file. Skipping.
在报错之后,应该是参考基因组的描述行没匹配上,而我却在参考基因组上修改,由于文件太大,每次修改都很慢,保存也慢。又要上传服务器运行,浪费了很多时间。以后就在一个小文件测试通过再看吧。记住啦~
后面怎么解决这个错误呢?
# 注释文件如下
chr1 12227 12594 ENSG00000223972.4 . + HAVANA intron . gene_id "ENSG00000223972.4"; transcript_id "ENSG00000223972.4"; gene_type "pseudogene"; gene_status "KNOWN"; gene_name "DDX11L1"; transcript_type "pseudogene"; transcript_status "KNOWN"; transcript_name "DDX11L1"; level 2; havana_gene "OTTHUMG00000000961.2";
chr1 12721 12974 ENSG00000223972.4 . + HAVANA intron . gene_id "ENSG00000223972.4"; transcript_id "ENSG00000223972.4"; gene_type "pseudogene"; gene_status "KNOWN"; gene_name "DDX11L1"; transcript_type "pseudogene"; transcript_status "KNOWN"; transcript_name "DDX11L1"; level 2; havana_gene "OTTHUMG00000000961.2";
chr1 13052 13220 ENSG00000223972.4 . + HAVANA intron . gene_id "ENSG00000223972.4"; transcript_id "ENSG00000223972.4"; gene_type "pseudogene"; gene_status "KNOWN"; gene_name "DDX11L1"; transcript_type "pseudogene"; transcript_status "KNOWN"; transcript_name "DDX11L1"; level 2; havana_gene "OTTHUMG00000000961.2";
# 本来参考基因组fna文件的描述行是下面这样
>NC_000001.10 Homo sapiens chromosome 1, GRCh37.p13 Primary Assembly
# 我尝试了 匹配不上第一次
>NC_000001.10 Homo sapiens chromosome 1, GRCh37.p13 Primary Assembly, chr1
# 我尝试了 匹配不上第二次
>chr1, NC_000001.10 Homo sapiens chromosome 1, GRCh37.p13 Primary Assembly
# 我尝试了 匹配不上第三次;这里也把注释文件改为chromosome 1
>chromosome 1
># 最终成功的是
>chr1
也就是说,描述行和bed注释文件完全一样才匹配上了。
bedtools getfasta -fi ncbi_dataset/data/GCF_000001405.25/human_ref_GRCh37.fna -bed introns.bed -fo introns.fasta
获取上下游位置和序列
这里获取序列的位置是以上述获得的
# 修改序列长度
# awk '{print $1"\t"($2-300)"\t"($3+300)}' tmp.bed > introns.bed
# -300是指向上游序列再取300 +300是指向下游序列再取300
# 这里有个问题,当$2-300时候,左边的序列长度居然是301
# 所以这里改为$2-299
awk '{print $1"\t"$7"\t""intron""\t"($2-299)"\t"($3+300)"\t"$5"\t"$6"\t"$9"\t"(substr($0, index($0,$10)))}' introns.bed > example_1.gtf
# 利用convert2bed包将gtf格式转化为bed格式
convert2bed -i gtf < example_1.gtf > example_1.bed
# 获取序列
bedtools getfasta -fi human_ref_GRCh37.fna -bed example_1.bed -fo example_1.fasta
这里虽然获取到了供体和受体对的序列,但是长度可能还会改变。而且还不能作为模型的输入,还有最后一步是提取供体和受体位点侧翼序列拼接在一起(例如文章DeepSplice中长度为120的四个部分序列)
暂时先记录到这里~
参考文章
链接: 下载参考基因组.
链接: 下载注释文件.
链接: UTR和CDS.
链接: bedtools文档.
链接: bedtools使用手册.
链接: bedtools常用命令.
链接: getfasta获取序列.
链接: GTF获取内含子位置.
链接: 获取内含子区域带图.
链接: 获取上下游序列.
链接: bedtools博客教程.
链接: bedtools博客教程2.
链接: DeepSplice.















 563
563











 被折叠的 条评论
为什么被折叠?
被折叠的 条评论
为什么被折叠?
 到【灌水乐园】发言
到【灌水乐园】发言









