









 本文介绍如何利用生物信息学工具进行基因功能注释,包括使用DIAMOND进行序列比对、GO和KEGG注释流程,以及利用R包GOstats进行GO和KEGG富集分析的方法。
本文介绍如何利用生物信息学工具进行基因功能注释,包括使用DIAMOND进行序列比对、GO和KEGG注释流程,以及利用R包GOstats进行GO和KEGG富集分析的方法。
准备
(1)用无参转录组分析软件得到unigene fasta文件,命名为my_unigenes.fa,格式如下表所示:
>MSTRG.5.1 gene=MSTRG.5
TGATGTCATCGATCCGTGACGTTTAGTATTCAACCAATAGGAATCAACCACGTAGGATTGGCGATCCTCG
TCAAACGTGTAAACGGTAGATCTGAACCGTTGACTGGCTGAGAGAAAACACATATTGTGGTATTTTAGTC
GGTACGATTAAGAAACACGAGAAACACACCGATGAGCGATAACAACCGTAGGATCGTCAACTTGCTTTTG
ACATCGTTGCCTATAAATTAAATATCAACAGCCCTACACGTGAGATTACTTTCATTATCTTCCCTTTTCG
AGATCGGAGCACTAATTTGAATTCAGAGAAGCGAAAGCGACGCAGAGAAGATGGCTCGTACCAAGCAGAC
CGCTCGTAAGTCTACCGGAGGAAAGGCCCCGAGGAAGCAGCTTGCTACTAAGGTATGGACTCTCTCTCTC
TCTCTCTCTCTCTCTCTCTCTCTC
>MSTRG.6.1 gene=MSTRG.6
GTAGAAATATAATGGGCTTAAAGATAAGGCCCATTAATTACAGAATCCTACGGGCACGTTACGTGCCGGT
TTAGTTTATTTTGCATTAAAAACCAATTTCGGAGTCGTCATCTTCCTCTTCGCCCAGATTCACTTCCTTC
GAGAAGATTCGCATCATTTTCCCAGTTATGGATTGGCAAGGACAGAAACTAGCGGAGCAGCTGATGCAGA
TCCTGCTCTTGATCGCCGCCGTTGTGGCGTTCGTCGTTGGTTACACGACGGCGTCGTTTAGGACGATGAT
GTTGATTTACGCGGGAGGGGTTGGTGTCACGACGTTGATCACGGTGCCGAACTGGCCATTCTTTAACCGT
CATCCACTCAAGTGGTTGGAACCAAGTGAAGCGGAGAAGCATCCTAAACCGGAAGTCGTTGTTAGCTCGA
AGAAGAAGTCATCTAAAAAGTAGCAAATCGTCTTTTGTAATCTCTCTTTTTTTTCTAGACCTTTGATATA
AAAAAAAAAGACATGTTCTGGATTTTGCTTATGAATAAGATAGTCTAAATGACATAATAATTTCGATTGA
TTCTGAGACATCCTTGCTTAATTGTTATGTA
>BnaA01g00010D.1 gene=BnaA01g00010D
TTTAGTTTATTTTGCATTAAAAACCAATTTCGGAGTCGTCATCTTCCTCTTCGCCCAGATTCACTTCCTT
CGAGAAGATTCGCATCATTTTCCCAGGTATAGATTCTCTGACGAGATCTGATTCTAGTTTGTTGCTTATT
GTTCTTGTAGATTCGAATCCGGCGATTATCAATTGCATTTCGTGCTGGATTCAATTCGAAAGATCCGATC
TAATCGTTTTGGTTGGTGTTGATTCAGTTATGGATTGGCAAGGACAGAAACTAGCGGAGCAGCTGATGCA
GATCCTGCTCTTGATCGCCGCCGTTGTGGCGTTCGTCGTTGGTTACACGACGGCGTCGTTTAGGACGATG
ATGTTGATTTACGCGGGAGGGGTTGGTGTCACGACGTTGATCACGGTGCCGAACTGGCCATTCTTTAACC
GTCATCCACTCAAGTGGTTGGAACCAAGTGAAGCGGAGAAGCATCCTAAACCGGAAGTCGTTGTTAGCTC
GAAGAAGAAGTCATCTAAAAAGTAG(2)下载并安装DIAMOND工具,可提升BLAST比对速度。
wget https://github.com/bbuchfink/diamond/releases/download/v0.9.18/diamond-linux64.tar.gz
tar xvzf diamond-linux64.tar.gz1.将unigenes比对到swissprot数据库(NR数据库同)
(1)获取swissprot数据库:
wget ftp://ftp.ncbi.nih.gov/blast/db/FASTA/swissprot.gz
gzip -d swissprot.gz
(2)建库:
diamond makedb --in swissprot -d swissprot
(3)blastx比对:
Evalue设定为1e-5,每query输出1条对应hit。阈值设定规则见参考2。
diamond blastx -d swissprot -q my_unigenes.fa -k 1 -e 0.00001 -o swiss_dia_matches.m8
得到的swiss_dia_matches.m8文件格式如下表所示,第二列为swissprot accession number:
MSTRG.5.1 Q5MYA4.3 96.2 26 1 0 331 408 1 26 7.6e-06 51.6
MSTRG.6.1 Q944J0.1 83.7 92 14 1 168 440 1 92 4.2e-36 152.5
BnaA01g00010D.1 Q944J0.1 83.7 92 14 1 240 512 1 92 1.4e-35 150.6
2.GO注释
(1)获取idmapping.tb.gz文件和Uniprot2GO_annotated.py文件(py文件下载见参考3):
wget ftp://ftp.pir.georgetown.edu/databases/idmapping/idmapping.tb.gzidmapping文件另一下载路径:
wget https://ftp.uniprot.org/pub/databases/uniprot/knowledgebase/idmapping/idmapping_selected.tab.gzidmapping.tb.gz文件格式如下表所示:
Q6GZX4 001R_FRG3G 2947773 YP_031579.1 81941549; 49237298 PF04947 GO:0006355; GO:0046782; GO:0006351 UniRef100_Q6GZX4 UniRef90_Q6GZX4 UniRef50_Q6GZX4 UPI00003B0FD4 654924 15165820 AY548484 AAT09660.1
Q6GZX3 002L_FRG3G 2947774 YP_031580.1 49237299; 81941548; 47060117 PF03003 GO:0033644; GO:0016021 UniRef100_Q6GZX3 UniRef90_Q6GZX3 UniRef50_Q6GZX3 UPI00003B0FD5 654924 15165820 AY548484 AAT09661.1
Q197F8 002R_IIV3 4156251 YP_654574.1 109287880; 123808694; 106073503 UniRef100_Q197F8 UniRef90_Q197F8 UniRef50_Q197F8 UPI0000D83464 345201 16912294 DQ643392 ABF82032.1
文件以tab键分隔,第1列为swissport accession number(Uniportkb ID),第4列为NR ID,第8列为GO注释。
(2)更改swiss_dia_matches.m8文件,将其第二列以“.”分开,只取“.”前面的字符串。更改后如下表所示:
MSTRG.5.1 Q5MYA4 96.2 26 1 0 331 408 1 26 7.6e-06 51.6
MSTRG.6.1 Q944J0 83.7 92 14 1 168 440 1 92 4.2e-36 152.5
BnaA01g00010D.1 Q944J0 83.7 92 14 1 240 512 1 92 1.4e-35 150.6
(3)修改下载的Uniprot2GO_annotated.py文件:
由idmapping.tb.gz文件格式可知:
为swissprot比对结果做GO注释时,第16行应改为:
UniProt_GO[lsplit[0]] = lsplit[7]为NR比对结果作GO注释时,第16行应改为:
UniProt_GO[lsplit[3]] = lsplit[7]此外,若在windows CMD中运行,第14行需加入decode()函数,linux中则不需要:
lsplit = line.decode().rstrip().split("\t")(4)运行py文件进行GO注释:
python UniProt2GO_annotate.py idmapping.tb.gz swiss_dia_matches.m8 swiss_go.outswiss_go.out文件格式如下表所示:
BnaA08g27970D GO:0045271,GO:0016021,GO:0005739,GO:0031966,GO:0009853,GO:0005747,GO:0055114
BnaAnng26910D GO:0031087,GO:0010606,GO:0000932,GO:0017148,GO:0042803,GO:0006397
BnaC07g50970D GO:0042938,GO:0042939,GO:0016021,GO:0042936,GO:0042937,GO:0022857,GO:0006807,GO:0005886,GO:0015031,GO:0009506
(5)用R包Org.Hs.eg.db为GO注释增添EVIDENCE注释:
用python将swiss_go.out文件的GO条目按“,”拆开,存入文件swiss_go_forStats:
def before_GOstat(f1,f2):
for i in f1.readlines():
j = i.split('\t')
for k in j[1].split(','):
m = j[0] + '\t' + k
if(m[-1] != '\n'):
m = m + '\n'
print(m)
f2.write(m)
f1 = open('swiss_go.out','r')
f2 = open('swiss_go_forStats','w')
f1.close()
f2.close()文件swiss_go_forStats格式如下表所示:
BnaA08g27970D GO:0005747
BnaA08g27970D GO:0055114
BnaAnng26910D GO:0031087
EVIDENCE注释:
>library(org.Hs.eg.db)
>#keytypes(org.Hs.eg.db)
>swiss_id <- read.delim('swiss_go_forStats',header = F)
>colnames(swiss_id) <- c('gene_id','GO')
>ev_id <- select(org.Hs.eg.db,keys = as.vector(swiss_id$GO),columns = c('EVIDENCE'),keytype = "GO")
>library(dplyr)
>swiss_goev <- left_join(swiss_id,ev_id[,1:2])
>write.table(swiss_goev,'swiss_goev_forStats',row.names = F,quote = F)
swiss_goev_forStats文件格式如下表所示:
gene_id GO EVIDENCE
BnaA08g27970D GO:0045271 NA
BnaA08g27970D GO:0016021 IDA
BnaA08g27970D GO:0016021 IBA
BnaA08g27970D GO:0016021 IEA
3.KEGG注释(以Brassica napus为例)
(1)KAAS网站自动注释my_unigenes.fa文件,得到基因名称对应的KO list,保存为all_kegg.txt文件,格式如下表所示:
MSTRG.6.1 K12946
BnaA01g00010D.1 K12946
BnaA01g00050D.1 K09338
(2)在http://www.genome.jp/kegg-bin/get_htext?htext=bna00001中找到物种名称,进入物种BRITE页面下载htext,得到bna00001.keg文件。
bna00001.keg文件格式如下:
+D GENES KO
#<h2><a href="/kegg/kegg2.html"><img src="/Fig/bget/kegg3.gif" align="middle" border=0></a> KEGG Orthology (KO) - Brassica napus (rape) </h2>
%<style type="text/css"><!-- #grid{ table-layout:fixed; font-family: monospace; position: relative; width: 1200px; color: black; }.col1{ position: relative; background : white; z-index: 1; width: 600px; overflow: hidden; }.col2{ position: relative; background : white; z-index: 2; padding-left: 10px; }--></style>
!
A<b>Metabolism</b>
B
B <b>Carbohydrate metabolism</b>
C 00010 Glycolysis / Gluconeogenesis [PATH:bna00010]
D 106359065 probable hexokinase-like 2 protein K00844 HK; hexokinase [EC:2.7.1.1]
D 106439029 hexokinase-3-like isoform X1 K00844 HK; hexokinase [EC:2.7.1.1]
D 106384641 probable hexokinase-like 2 protein K00844 HK; hexokinase [EC:2.7.1.1]
(3)python解析bna00001.keg文件,得到path id(C)对应的KO号(D K*****),为all_kegg.txt文件注释path id,结果保存在all_kegg_path文件中。
import re
f1 = open('bna00001.keg','r')
dict1 = {}
dict2 = {}
for i in f1.readlines():
if(i[0] == 'C'):
d = re.search('\d+',i)
if(i[0] == 'D'):
h = re.search('K\d+',i)
dict2[h.group()] = d.group()
dict1.setdefault(d.group(), set()).add(h.group())
f1.close()
f2 = open('all_kegg.txt','r')
f3 = open('all_kegg_path','w')
for j in f2.readlines():
jj = j.split('\t')[1].split('\n')[0]
for k,v in dict1.items():
#print(v)
if(jj in v):
u = j.split('\n')[0]+'\t'+ k + '\n'
print(u)
f3.write(u)
f2.close()
f3.close()all_kegg_path文件格式如下表所示:
MSTRG.6.1 K12946 03060
BnaA01g00010D.1 K12946 03060
MSTRG.16.1 K02266 00190
4.用R包GOstats进行GO及KEGG富集分析
(1)安装GOstats
>source("https://bioconductor.org/biocLite.R")
>biocLite("GOstats")(2)GO富集分析(以CC为例,BP、MF同)
>library(GOstats)
>ago <- read.delim('swiss_goev_forStats',row.names = NULL)
>colnames(ago) <- c('gene_id','go_id','evi')
>goframeData <- na.omit(data.frame(ago$go_id,ago$evi,ago$gene_id))
>goFrame <- GOFrame(goframeData)
>goAllFrame <- GOAllFrame(goFrame)
>library('GSEABase')
>gsc <- GeneSetCollection(goAllFrame,setType = GOCollection())
>universe <- as.vector(ago$gene_id)
>DEGfile <- read.csv('my_degs.csv',header = T,row.names = NULL)
>genes = as.vector(DEGfile$gene_id)
>params <- GSEAGOHyperGParams(name = 'custom',
geneSetCollection = gsc,
geneIds = genes,
universeGeneIds = universe,
ontology = 'CC',
pvalueCutoff = 0.05,
conditional = FALSE,
testDirection = 'over')
>Over <- hyperGTest(params)
>write.csv(summary(Over),'GO_CC_hyper.csv')
GO_CC_hyper.csv格式如下表所示:
"","GOCCID","Pvalue","OddsRatio","ExpCount","Count","Size","Term"
"1","GO:0005737",9.49537738427155e-10,1.31027637565836,1072.90234330174,1208,45123,"cytoplasm"
"2","GO:0044444",2.6104214493046e-06,1.22794626065899,835.462394712079,937,35137,"cytoplasmic part"
"3","GO:0044424",5.37119511677146e-06,1.27132246803014,1571.3455279888,1655,66086,"intracellular part"
(3)KEGG富集分析
>library(GOstats)
>ago <- read.delim('all_kegg_path',header = F,row.names = NULL)
>colnames(ago) <- c('gene_id','KO_id','kegg_id')
>ago$kegg_id <- sprintf("%05d",ago$kegg_id)
>keggframeData <- na.omit(data.frame(ago$kegg_id,ago$gene_id))
>keggframeData <- unique(keggframeData)
>keggFrame <- KEGGFrame(keggframeData)
>library('GSEABase')
>gsc <- GeneSetCollection(keggFrame,setType = KEGGCollection())
>universe <- as.vector(ago$gene_id)
>DEGfile <- read.csv('my_degs.csv',header = T,row.names = NULL)
>genes <- as.vector(DEGfile$gene_id)
>params <- GSEAKEGGHyperGParams(name = 'custom',
geneSetCollection = gsc,
geneIds = genes,
universeGeneIds = universe,
pvalueCutoff = 0.05,
testDirection = 'over')
>Over <- hyperGTest(params)
>write.csv(summary(Over),'KEGG_hyper.csv')
(4)ggplot2作富集term条形图(以GO富集分析结果为例)
>library(ggplot2)
#选择颜色
>library(RColorBrewer)
>display.brewer.all()
>col3 <- brewer.pal(4,'Set3')[c(1,3,4)]
>cc = read.csv('GO_CC_hyper.csv',header = T,row.names = NULL)
>bp = read.csv('GO_BP_hyper.csv',header = T,row.names = NULL)
>mf = read.csv('GO_MF_hyper.csv',header = T,row.names = NULL)
#设定作图阈值为Pvalue<1e-9
>cc2 <- cc[cc$Pvalue<1e-9,]
>bp2 <- bp[bp$Pvalue<1e-9,]
>mf2 <- mf[mf$Pvalue<1e-9,]
>colnames(cc2)[2] <- c('GOID')
>colnames(bp2)[2] <- c('GOID')
>colnames(mf2)[2] <- c('GOID')
>data <- rbind(mf2,cc2)
>data <- rbind(data,bp2)
>data2 <- data.frame(data,type = c(rep('MF',dim(mf2)[1]),rep('CC',dim(cc2)[1]),rep('BP',dim(bp2)[1])))
>p <- ggplot(data2,aes(x = Term,y = log2(Count),fill = type,order = type))+
geom_bar(stat = 'identity')+coord_flip()+
scale_fill_manual(values = col3,limits = c('BP','CC','MF'),labels = c('BP','CC','MF'))+
theme_bw()+theme(legend.title=element_blank(),
axis.text = element_text(size = mysize),
axis.title.x = element_text(size = rel(1.5)),
axis.title.y = element_text(size = rel(1.5),angle = 90),
legend.text = element_text(size = rel(1.5)))+
ylab('Number of Genes (log2)') + xlab('Terms')
>ggsave('go_hyper.tiff',p,device = 'tiff',scale = 1,width = 20,height = 15,dpi = 300)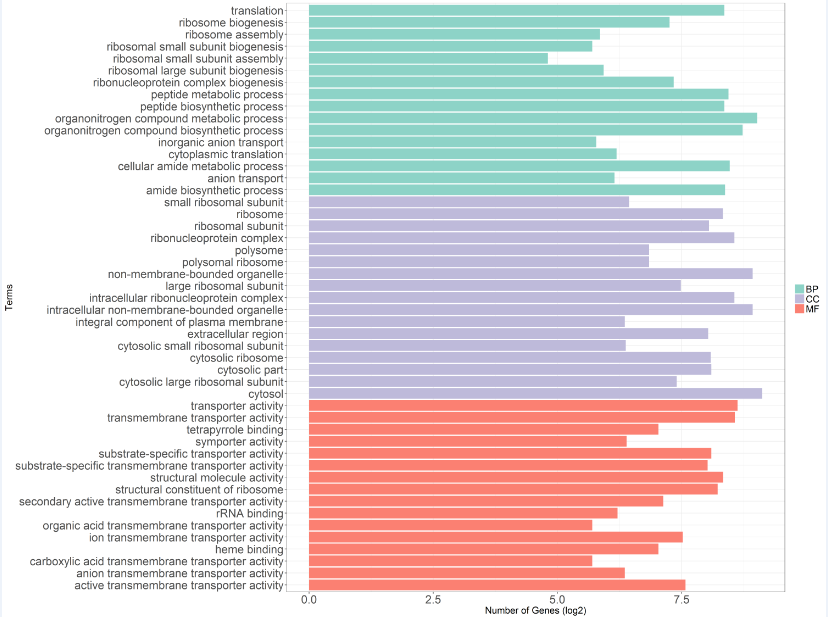
参考
6.GOstats-Hypergeometric tests for less common model organisms


















 被折叠的 条评论
为什么被折叠?
被折叠的 条评论
为什么被折叠?
 到【灌水乐园】发言
到【灌水乐园】发言









