









A knowledge graph to interpret clinical
proteomics data
Alberto Santos 1,2,3 ✉, Ana R. Colaço1
, Annelaura B. Nielsen1
, Lili Niu1
, Maximilian Strauss1,4
,
Philipp E. Geyer 1,4,5
, Fabian Coscia 1,5
, Nicolai J. Wewer Albrechtsen 1,6,7
, Filip Mundt1
https://doi.org/10.1038/s41587-021-01145-6
【一共8页】
笔记
本文介绍了临床知识图谱(CKG)这一开源平台,它整合多源数据构建近2000万个节点和2.2亿条关系的图数据库,通过分析核心模块、图数据库构建及报告管理器等功能,在非酒精性脂肪肝、癌症等疾病研究中加速数据解析、辅助临床决策,推动精准医学发展 。
论文 “A knowledge graph to interpret clinical proteomics data” 发表于Nature Biotechnology,介绍了临床知识图谱(CKG)这一开源平台,旨在整合多源数据,解决临床蛋白质组学数据分析和解释难题,推动精准医学发展。
- 问题:精准医学需整合组学数据辅助临床决策,但生物医学数据量多且杂,相关知识分散于多个数据库和文献中,数据集成困难;现有蛋白质组学工作流程难以处理日益增长的数据量,高通量蛋白质组学定量结果的解读也存在瓶颈。
- 挑战:生物医学数据多样、异构且分布在多个平台,难以协调和整合;大量科学数据和知识存在于未标准化的期刊出版物中;现有蛋白质组学工作流程难以适应数据量增长,缺乏有效解读定量结果的工具。
- 创新点:构建 CKG 开源平台,整合实验数据、公共数据库和文献,以图结构提供灵活数据模型;融入统计和机器学习算法,加速蛋白质组学工作流分析与解释;利用 Jupyter notebooks 实现可重复、可再现和透明的分析。
- 贡献:为蛋白质组学和多组学数据分析提供统一框架;通过案例研究展示其在疾病生物标志物发现、治疗方案优先级排序等方面的应用;促进科学研究的可重复性,推动精准医学发展。
- 提出的方法:开发 CKG 平台,包含分析核心模块(用于数据处理、分析和可视化)、图数据库构建模块(整合多源数据构建图数据库)、图数据库连接模块(连接和查询数据库)和报告管理器模块(生成分析报告);采用 Python 及相关科学库进行开发,兼容多种数据格式。
- 指标:通过分析蛋白质组学数据的定量指标,如蛋白质表达量变化、变异系数等,结合统计检验(如 ANOVA、t 检验)确定差异显著性;利用富集分析挖掘功能信息,以评估疾病相关通路和生物学过程的变化。
- 模型结构:CKG 平台采用模块化架构,各模块独立又协同工作。图数据库以 Neo4j 为后端,定义 36 种节点标签和 47 种关系类型,连接多层面临床蛋白质组学实验数据并注释;分析核心模块涵盖数据处理、探索、分析和可视化等功能。
- 结论:CKG 是一个开放、强大的框架,可实现蛋白质组学和多组学数据的透明、自动化和集成分析,满足可重复性科学研究的需求,有助于解决个性化医疗和临床决策过程中的关键瓶颈问题。
- 剩余挑战和未来工作:需进一步优化以处理大规模数据;探索与人工智能技术的深度融合,如利用图深度学习提升性能;加强数据隐私保护,通过联邦学习等技术在保护数据隐私的同时实现跨平台分析。
- 数据集:使用多个蛋白质组学数据集,包括来自 PRIDE 数据库的非酒精性脂肪肝(PXD011839)、癌症相关研究(PXD008713、PXD010372)等;还使用了临床蛋白质组肿瘤分析联盟(NCI/NIH)提供的胶质母细胞瘤数据,以及马萨诸塞州总医院的 Olink 蛋白质组学数据。
正文
【全文介绍】
实施精准医学取决于将蛋白质组学等组学数据整合到临床决策过程中,但生物医学数据的数量和多样性,以及临床相关知识在多个生物医学数据库和出版物中的传播,对数据集成构成了挑战。在这里,我们介绍了临床知识图谱 (CKG),这是一个开源平台,目前包含近 2000 万个节点和 2.2 亿个关系,代表相关的实验数据、公共数据库和文献。图形结构提供了一个灵活的数据模型,当新数据库可用时,该模型可以轻松扩展到新的节点和关系。CKG 结合了统计和机器学习算法,可加速典型蛋白质组学工作流程的分析和解释。使用一组概念验证生物标志物研究,我们展示了 CKG 如何增强和丰富蛋白质组学数据,并帮助为临床决策提供信息。
循证精准医学的范式已经朝着更全面的疾病表型分析发展。这需要无缝集成各种数据,notypes。这需要无缝集成各种数据,例如临床、实验室、成像和多组学数据(基因组学、转录组学、蛋白质组学或代谢组学)1。最近,我们发现,结合临床和分子数据的更精细的疾病定义可以更深入地了解个体的疾病表型并揭示候选标志物
预后和 / 或治疗 2-4。此外,多组学数据可以生成新的假设,最终转化为临床上可行的结果 5。生物医学研究界很早就认识到需要收集、组织和构建相关数据,因此在社区范围内采用了多个生物医学数据库(补充表 1)。然而,协调和集成仍然具有挑战性,因为它通常是多样化的、异构的,并且分布在多个平台上。此外,许多科学数据和知识只 “存储” 在数百万非标准化期刊出版物中。
在过去十年中,基于质谱 (MS) 的蛋白质组学取得了长足的进步,现在提供了越来越全面的生物过程、细胞信号转导事件和蛋白质相互作用的视图 6。然而,目前使用的基于 MS 的蛋白质组学工作流程是在十多年前就被概念化的,快速增长的数据量给该领域带来了新的挑战。高通量蛋白质组学中一个更大且不断增长的瓶颈是难以解释定量结果以形成生物学或临床假设。只有少数工具旨在缓解这个问题 7,8 需要集成多种数据类型的解决方案,同时捕获分子实体与由此产生的疾病表型之间的关系。此外,我们看到对更具包容性的解决方案的需求日益增长,这些解决方案为那些缺乏专业知识的人提供
以更用户友好的方式从蛋白质组学数据中提取高质量信息。因此,将一系列数据库和科学文献信息与组学数据集成到易于使用的工作流程中的基于知识的平台将增强发现科学和临床实践的能力。
网络和图形已成为自然的代表方式发送连接数据,也包括生物学 9-11。期间的努力
在过去的十年中,组织了大量不同的信息。tion 作为节点(实体)和边缘(关系)的集合 12-16。
由此产生的灵活结构(称为知识图谱)可以快速适应具有关系的复杂数据,并支持有效使用网络分析技术来识别隐藏的模式和知识 13,17-19。
在这里,我们将这个概念带入一个新的方向,并描述了一个知识图谱框架,该框架有助于蛋白质组学与其他组学数据的协调,同时整合相关的生物医学数据库和从科学出版物中提取的文本。它被称为 CKG,构成了一个包含数百万个节点和关系的图形数据库。它允许具有临床意义的查询和高级统计分析,从而实现自动化数据分析、知识挖掘和可视化。CKG 通过构建科学的 Python 库 20 来整合社区工作,这也使平台可靠、可维护且不断改进。整个系统是开源的,并且具有宽松的许可(通过 MIT 许可证)。它支持在标准工作流程和基于 Jupyter Notebook 的交互式探索中进行可重复、可重现和透明的分析。
结果
CKG 架构概述。CKG 包括几个独立的功能模块,用于 (1) 格式化和分析蛋白质组学数据 (analytics_core;(2) 构建一个图数据库通过整合来自一系列可公开访问的数据库、用户进行的实验、现有本体和科学出版物的可用数据 (graphdb_builder);(3) 连接并查询此图数据库 (graphdb_connector);(4) 通过在线报告 (report_manager) 和 Jupyter 笔记本促进数据可视化、存储库和分析(图 1a、b)。此架构可无缝协调和集成数据以及用户提供的分析。它还有助于数据共享和可视化以及基于生物医学知识注释的详细统计报告的解释,从而生成临床相关的结果。在接下来的几节中,我们将介绍各个模块和知识图谱构建过程。
分析核心作为开放式蛋白质组学分析框架。蛋白质组学数据下游分析的第一步需要全面且通用的统计、机器学习和可视化方法。MSstats 和 Perseus 通过提供多功能统计和生物信息学工具来分析基于 MS 的定量蛋白质组学,从而推进蛋白质组学数据 7,8。以这些和其他努力为参考,我们开发了
CKG 的分析核心,以透明和高效的方式包含所需的功能。我们选择 Python 及其相关的科学堆栈,因为这使我们能够采用经过充分测试和最新的算法,同时避免重新实现现有的方法。分析核心中实现的功能以统计和可视化数据表示为中心,涵盖所有主要计算领域,例如表达、交互和翻译后、基于修饰的蛋白质组学(图 1b)。
我们设计的分析核心包括数据科学管道中的主要步骤:数据准备(过滤、标准化、插补和数据格式化)、数据探索(汇总统计、排名和分布)、数据分析(降维、假设检验和相关性)和可视化。除了蛋白质组学之外,该分析核心还集成了对其他数据类型(即临床数据、多组学、生物背景和文本挖掘)的分析,从而超越了以前的工作。此外,为了补充广泛的 Python 产品组合,我们整合了以 R 语言(即 SAMR)优化的函数和 WGCNA21-23)(补充表 2)。
我们提供了一个可视化模块(可视化),该模块使用与 Python 和 R 兼容的图形库 Plot.ly (Data Apps for Production | Plotly) 涵盖基本绘图(例如,条形图或散点图)和更复杂的绘图(例如,网络、桑基或极坐标图)。这样,由 CKG 框架创建的可视化可以很容易地从其他语言(方法)导出和使用。
由于其模块化设计,分析核心可以在 CKG 框架内使用,也可以通过从 Python 导入来独立使用。同样,分析和可视化功能不仅限于蛋白质组学数据,还可以处理矩阵格式的任何类型的数据。开放式设计促进了新分析方法和可视化的轻松集成。我们集成了 Jupyter Notebooks,这是一种开源工具,允许在单个文档中混合文本、图形、代码和数据 24-27,支持标准或定制的分析管道,包括添加Python 或 R 生态系统中的现有或用户实现的功能(方法)。
构建和填充图形数据库。为了实现蛋白质组学数据与其他组学实验和 / 或文献信息的无缝注释和集成,我们构建了一个自然连接大型异构数据的图形数据库(补充图 1a)。我们选择开源的 Neo4J 数据库平台作为我们当前的后端,因为它的性能、行业接受度和相关的 Cypher 查询语言 (https://neo4j. com/) (补充图 1b)。为了构建知识图谱,我们首先编写了一个解析器库 (graphdb_builder),其中包含每个本体、数据库和实验类型的相关配置。这些解析器从在线资源下载数据,提取信息并生成实体(节点)和关系,这两者都可以在蛋白质节点中具有属性,例如名称或描述。解析器使用成对的配置文件,这些文件指定需要如何解释本体、数据库或实验。这种设计允许不受限制地集成新资源或加工工具。它们的输出格式往往会不时更改,但这只影响一个可以轻松适应的解析器 / 配置。例如,当前 CKG 的蛋白质组学解析器接受来自常用程序(如 MaxQuant、Spectronaut、
FragPipe 或 DIA-NN28–31,或社区标准格式 mzTab
基于质谱的蛋白质组学和代谢组学数据 32。当新的处理程序出现时,可以很容易地扩展 CKG 以接受额外的数据输出。
一旦本体、数据库和实验文件被标准化、格式化和导入,graphdb_builder 模块就会将它们加载到图形数据库中,其中包含一组 Cypher 查询,这些查询会创建相应的节点和关系(方法)。我们的数据模型将 36 个不同的节点标签与 47 种不同的关系类型连接起来(图 1c)。为了制作实验蛋白质组学数据 33,我们设计了一个数据模型,能够支持围绕每个研究项目存储标准化元数据(例如研究的疾病和干预措施),为注册个体、收集的生物样本和分析样本定义唯一标识符(补充图 2a)。它支持对实验确定的蛋白质命中进行预定义查询,关于它们与所研究的疾病(本体论关联)、药物或注释的基因本体术语和通路的关联。这些类型的查询提供了对改变功能的见解,建议用于调节蛋白质的药物和与代谢物的联系,以揭示可能的混杂因素。
CKG 包括数百万个节点和关系。CKG 数据库持续增长,目前使用 10 个本体从 26 个生物医学数据库中收集注释,并将这些信息组织成近 2000 万个节点,由 2.2 亿个关系连接(补充图 1a)。其中超过 5000 万个关系涉及 “出版物” 节点,这些节点将有关人体系统研究的科学出版物(用 PubMed 标识符编码)与蛋白质、药物、疾病、功能区域和组织联系起来(补充图 3a)。它们是衍生的
来自近 700 万篇摘要和全文文章的命名实体识别(占总出版物的 8.5% 基于全文,但
20.4% 来自过去 10 年)34,35,从而概括了
在同行评审的出版物中积累了生物医学知识。
我们发现图形结构易于扩展,有助于集成新的本体、数据库和实验。此外,这种固有的灵活性允许最初设计为主要为大规模蛋白质组学数据解释提供生物医学背景的节点和关系易于建模以集成其他组学数据集。例如,为了整合代谢组学数据,我们可以将数据库中已经存在的代谢物节点与分析样品联系起来。将代谢物与节点(如通路、蛋白质、组织或疾病)连接起来的现有关系有助于解释并与蛋白质组学结果无缝集成。
CKG 框架提供了一个基础设施,有助于利用图中的现有连接以及 Neo4j 和 Python 库(如 NetworkX36)中已经实现和优化的图算法。例如,当集成新项目时,默认分析可以在图表中识别类似的项目,并且这些比较的结果将显示在项目报告中。此功能根据已识别蛋白质的重叠(Jaccard 和重叠相似性)或类似蛋白质谱(Pearson 相关性)来比较项目。此外,CKG 还为网络分析和机器学习算法的应用提供了一个框架。学习图结构
使用图表示学习 37,38 可以增强预测
的新链接,一种称为 Link Prediction 或 Graph Comple 的策略
tion39,40 (方法).
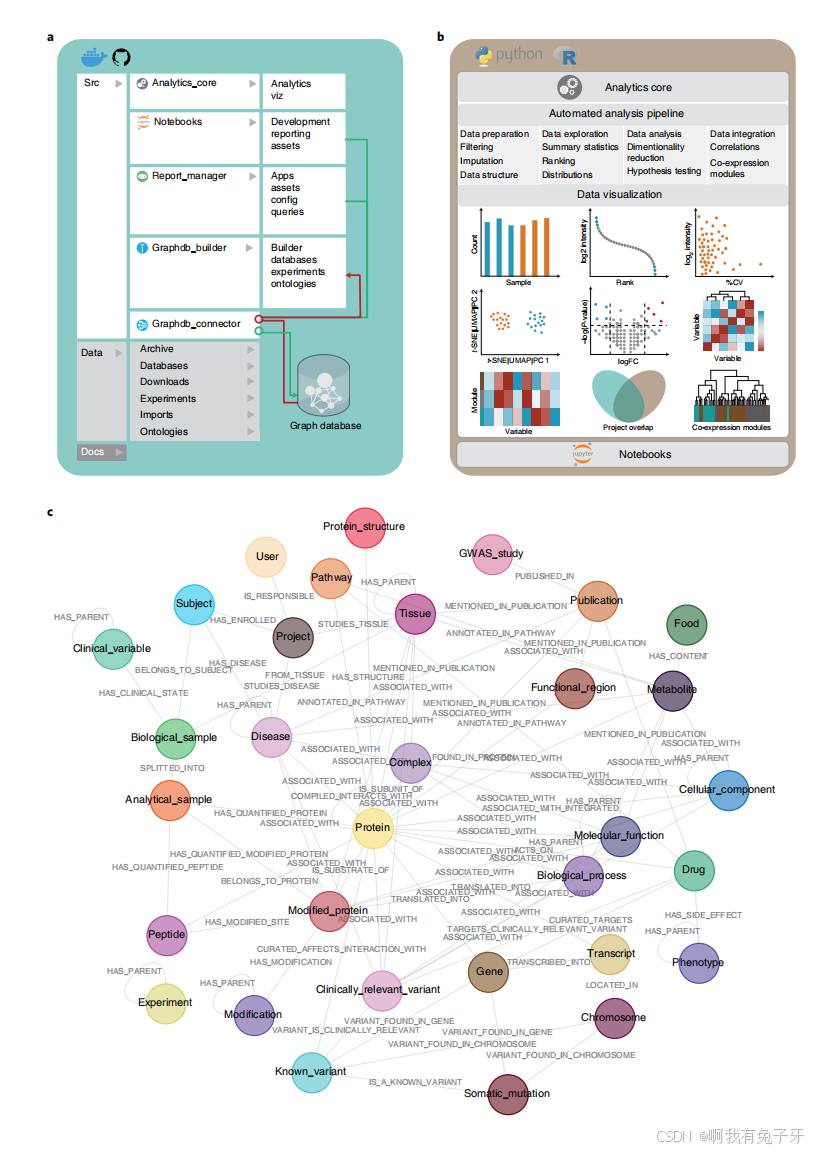
{图 1 | 临床知识图谱架构。a, CKG 架构是用 Python 实现的,包含几个独立的模块,分别负责连接图数据库 (graphdb_connector)、构建图 (graphdb_builder)、分析和可视化实验数据 (analytics_core)、显示和启动多个应用程序 (report_manager); 它还包含带有分析示例 (notebook) 的 Jupyter 笔记本存储库。该代码可通过 https://github.com/MannLabs/CKG 访问,也可以作为完整的 Docker 容器访问。b, CKG 分析核心实现了多种最新的数据科学算法,用于蛋白质组学数据的统计分析和可视化:数据准备、探索、分析和可视化。该库还可以直接在 Jupyter 笔记本中使用,独立于其他 CKG 模块,并用于分析其他组学类型。c,CKG 图数据库数据模型旨在整合多层次临床蛋白质组学实验,并用生物医学数据对其进行注释。它定义了不同的节点(例如,蛋白质、代谢物和疾病)以及连接它们的关系类型(例如,HAS_PARENT 和 HAS_QUANTIFIED_PROTEIN)。FC, 换折;Src,源代码。}
提取可作知识的框架。CKG 的一个主要目标是将分析模块的强大功能与
将大量先验信息集成到图形数据库中,以最好地解释基于 MS 的蛋白质组学或其他组学实验。这些异构但相互连接的数据源的协调使标准分析管道能够自动报告结果,以更一致的格式取代数周的手动工作。这些标准报告提供了对所生成数据质量的初步评估,突出显示了相关命中,并将这些命中与图表中的不同生物医学成分联系起来。Report Manager 组件 (report_manager) 协调创建和更新实验项目以及自动分析、可视化和知识提取(图 2)。
报告管理器是作为仪表板应用程序的集合实现的,这些应用程序与数据库连接,用于概述知识图谱 (Home)、创建和上传新的临床蛋白质组学项目(项目创建和数据上传)以及运行自动分析管道 (ProjectApp)。这定义了从项目想法到基于知识的分析报告的工作流程(补充图 2b)。项目创建和数据上传步骤在 CKG 中为项目、登记的队列个体(如果适用)、收集的生物样品以及通过基于 MS 的蛋白质组学分析的分析样品生成节点和唯一标识符。这还包括与疾病、组织和临床干预的联系(补充图 2c),它们提供先验知识,例如与这些疾病相关的已知蛋白质以及与这些疾病相关的组织或文献。一旦临床和 / 或蛋白质组学数据准备好并处理,它们就会通过 “数据上传” 仪表板应用程序集成到图表中(补充图 2d)。
上传数据会触发生成器的导入和加载过程,并在新项目中生成所有必要的关系,包括项目中量化的蛋白质、肽和蛋白质修饰的链接。报表使用新链接
Manager 模块来分析与项目相关的不同数据类型。这些分析是使用配置文件预定义的,这些文件以标准化、灵活和可扩展的方式分解了整个分析工作流程的步骤。它们描述了输入数据、要执行的分析、要使用的参数以及如何可视化结果。然后,最终报告是为每种数据类型创建的可视化序列,这些可视化被划分到仪表板中的不同选项卡(绘图和表格)。
除了可在浏览器中查看之外,所有报告、分析结果和可视化都可以下载为单个压缩文件,其中包含可立即发布的表格和图形。此外,它们还提供分层数据格式 (HDF5),这是一种受许多编程语言支持的标准且可扩展的文件格式,从而实现互作性。这种设计有助于新开发的分析和可视化的持续集成(补充图 4)。此外,可以共享配置文件,这提高了透明度并促进了可复制性和再现性。
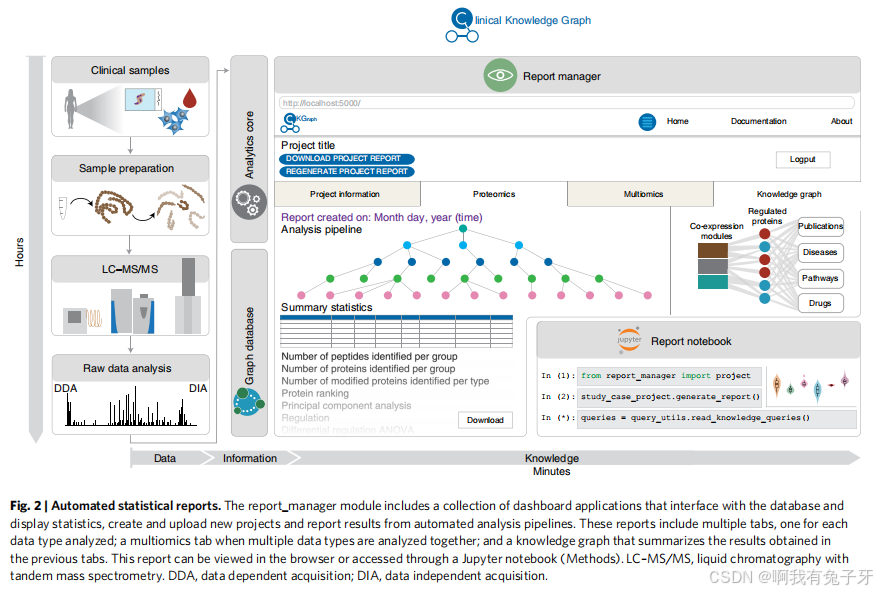
{ 2 | 自动统计报告。report_manager 模块包括一组仪表板应用程序,这些应用程序与数据库交互并显示统计数据,创建和上传新项目以及报告来自自动分析管道的结果。这些报表包括多个选项卡,每个选项卡对应分析的每种数据类型;当一起分析多种数据类型时,多组学选项卡;以及一个知识图谱,用于汇总在前面的选项卡中获得的结果。此报表可以在浏览器中查看,也可以通过 Jupyter 笔记本(方法)访问。LC-MS/MS,液相色谱 - 串联质谱。DDA,数据依赖性采集;DIA,数据非依赖型采集。}
用于发现肝脏疾病生物标志物的自动化 CKG 分析。为了展示 CKG 如何加速和扩展数据的分析和解释,我们在非酒精性脂肪性肝病 (NAFLD) 的蛋白质组学研究中使用了其默认管道 41(图 3)。
对于临床数据,CKG 默认分析管道自动总结了队列的临床特征,并突出显示了研究组之间具有显着差异的变量(健康和正常葡萄糖耐量 (NGT)、2 型糖尿病 (T2D)、NGT 伴 NAFLD 和 T2D 伴 NAFLD 和肝硬化)。这证实了肝酶丙氨酸氨基转移酶、天冬氨酸氨基转移酶和碱性磷酸酶测量或血红蛋白 A1c (HbA1c) 等水平的显着差异。
蛋白质组学默认分析从鉴定的肽和蛋白质的概述以及蛋白质组学数据矩阵的描述性统计的详细摘要开始。CKG 继续对蛋白质组覆盖度、动态范围、样品中的蛋白质变异系数 (CV) 和基于已知组织质量标志物的样品质量控制进行可视化 42。这些排名使用从知识图谱中挖掘的精选信息自动进行注释,在这种情况下,突出显示了临床中已经使用的与 NAFLD 或肝硬化相关的标志物(补充图 5)。
默认数据分析使用主成分分析来降低特征的维数,以便获得数据概览。然后,使用事后检验的方差分析确定所有研究组之间和特定组对之间的统计学显着差异(事后分析)。事后测试以交互式火山图的形式呈现,其中包含有关具有预定义显着性阈值的上调和下调蛋白质的信息(即,折叠 change>2 和错误发现率 (FDR)<0.05(图 3a)。CKG 自动重现了我们之前的结果,显示参与免疫系统调节和炎症的蛋白质失调,例如 C7、JCHAIN、PIGR 和 A2M,这是一种已知的肝纤维化标志物,CKG 报告了 14 篇出版物,证实了这种联系。此外,CKG 强调与健康个体相比,肝硬化患者的 TTR-RBP 复合物 (TTR 和 RBP4) 下调。该复合物参与类视黄醇代谢,其失调与肝脏疾病和细胞外基质沉积的改变有关,导致纤维化 43。此外,该报告揭示了 CD5L 与肝硬化、肝细胞癌和其他肝病的调节作用之间的文献和数据库关联 44。这些代谢上有趣的发现在我们的手动分析中被遗漏了,但 CKG 的自动化管道优先考虑了这些发现,该管道提取了不同条件下显着调节的蛋白质。为了将相关的蛋白质变化可视化为一个网络,
默认分析将具有显著关联的蛋白质(Pearson 相关系数 >0.5 和 FDR<0.05)联系起来。使用鲁汶算法检测高度相关的蛋白质簇 45 揭示了潜在的临床相关联系,例如由 PIGR 组成的簇以及 DPP4 和 TGFBI 与肝纤维化的关联。数百万种蛋白质相互作用的背景知识使 CKG 能够识别出六个主要簇,这些簇将细胞外基质重塑因子、互补成分和炎症标志物组合在一起,它们连接了两个候选生物标志物(PIGR 和 JCHAIN)。
将差异调节的蛋白质与药物、疾病和出版物以及丰富的生物过程和途径相关联,确定了 NAFLD 中其他失调的途径,这些途径在我们之前的分析中被忽视了。这些研究包括 “胰岛素样生长因子 (IGF-1) 转运的调节” 和 “胰岛素样生长因子结合蛋白 (IGFBP) 的摄取”,这与 IGFBP3 酸不稳定亚基 (IGFALS) 的变化有关。值得注意的是,最近对此类关联进行了调查
NAFLD46,47 的因果关系和治疗潜力。
CKG 还报告了蛋白质与疾病的关联,表明可能与肝癌、肝炎和胰腺疾病共享疾病机制。
项目中存在各种数据类型触发了默认的多组学分析管道。在全球临床蛋白质组学相关性分析 48 中,临床肝酶值与 HbA1c、空腹血糖水平以及肝纤维化和肝硬化的几种候选生物标志物(如 PIGR、TGFBI、ANPEP 和 C7)聚集在一起(图 3b)。CKG 还使用 WGCNA 获得共表达蛋白的模块,而不是与临床变量相关的单个蛋白(图 3c)。
最后,自动分析管道将所有临床、蛋白质组学和多组学分析总结为一张图表,其中包含所有受监管的蛋白质以及从知识图谱中提取的关系(例如疾病、药物、相互作用和途径),使用中介中心性来确定优先级并减少呈现的节点数量 (Fig. 3d)。
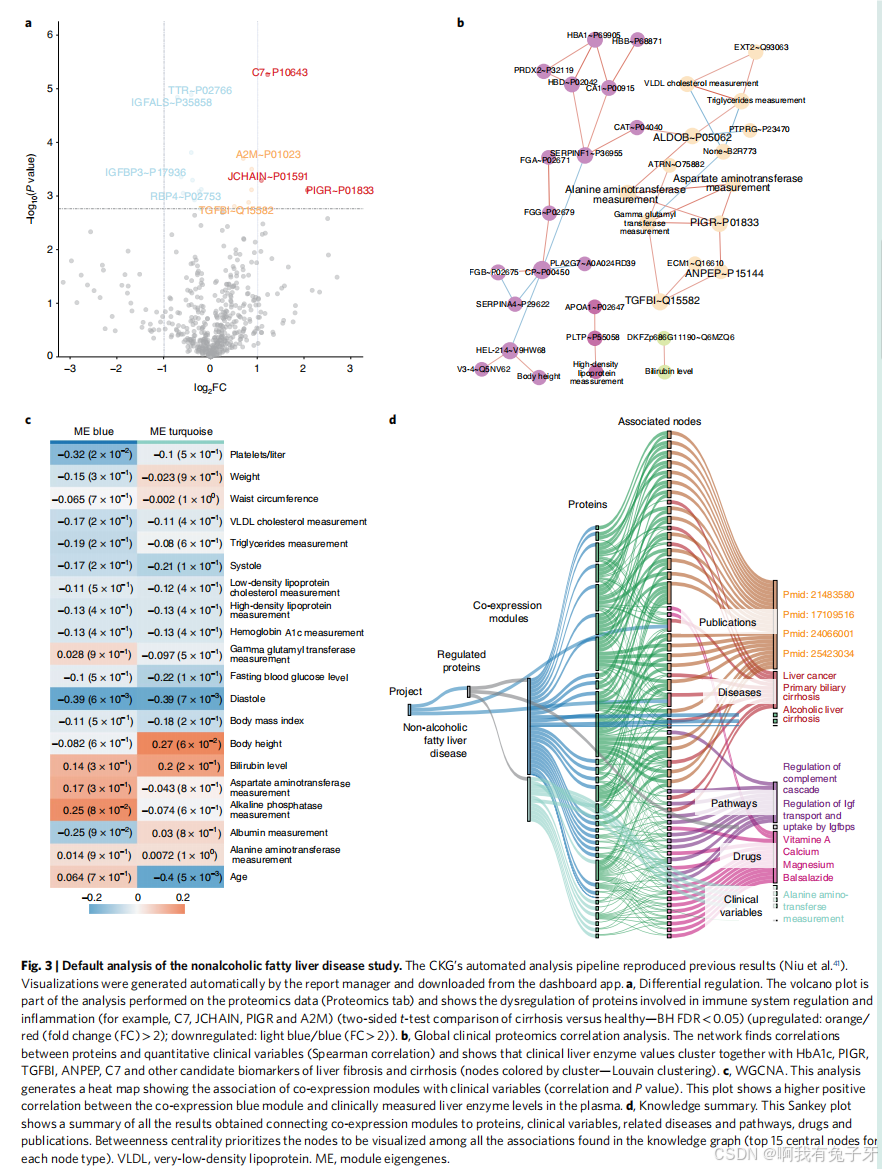
{图 3 | 非酒精性脂肪性肝病研究的默认分析。CKG 的自动分析流程重现了以前的结果(Niu et al.41)。可视化由报告管理器自动生成,并从仪表板应用程序下载。a,差分调节。火山图是对蛋白质组学数据(蛋白质组学选项卡)进行的分析的一部分,显示了参与免疫系统调节和炎症的蛋白质(例如,C7、JCHAIN、PIGR 和 A2M)的失调(肝硬化与健康的双侧 t 检验比较 - −BHFDR<0.05(上调:橙色 / 红色(倍数变化 (FC)>2) :d ownregulated:浅蓝色 / 蓝色 (FC>2) b,全球临床蛋白质组学相关分析。该网络发现蛋白质与定量临床变量(Spearman 相关性)之间的相关性,并显示临床肝酶值与 HbA1c、PIGR、TGFBI、ANPEP、C7 和其他肝纤维化和肝硬化的候选生物标志物(节点由聚类着色 - 鲁汶聚类)聚集在一起。c, WGCNA. 该分析生成一个热图,显示共表达模块与临床变量(相关性和 P 值)的关联。该图显示共表达蓝色模块与临床测量的血浆中肝酶水平之间存在较高的正相关性。d、知识总结。该 Sankey 图显示了将共表达模块与蛋白质、临床变量、相关疾病和通路、药物和出版物联系起来获得的所有结果的摘要。中介中心性在知识图谱中找到的所有关联(每种节点类型的前 15 个中心节点)中优先考虑要可视化的节点。VLDL,极低密度脂蛋白。ME,模块特征基因。}
整个默认管道只用了不到 5 分钟,但基本上捕获了我们从之前的手动分析中收集的所有见解,而我们之前的手动分析需要数周时间。对差异丰度蛋白质的解释包括耗时的文献和数据库搜索已知 / 已发表的蛋白质 - 疾病关联和知识收集 41,但 CKG 显示它们是不完整的。
CKG 支持多蛋白质组学数据集成,用于癌症生物标志物的发现和验证。为了探索 CKG 的多分析能力,我们重新分析了最近的一项研究,在该研究中,我们将癌症 / 睾丸抗原家族 45 (CT45) 确定为卵巢浆液腺癌长期生存的生物标志物,并描述了其作用方式 3。多维蛋白质组学、磷酸化蛋白质组学和互通组学在 CKG 中被建模为不同的关联项目,并使用适用于每种数据类型(蛋白质组学、互溶组学和磷酸化蛋白质组学)的默认分析进行独立分析。CKG 再现的 CT45 在化疗后长期缓解的患者中表达显著更高(图 4a、b)。CKG 还证实,以前几乎没有关于 CT45 的细胞作用和功能的知识,但产生了 CT45 的 24 个潜在相互作用子,其中 4 个属于 PP4 复合物,由人类相互作用图 49 贡献(图 4c)。
PP4 复合物与 DNA 损伤修复有关 50,再加上患者接受了化疗诱导 DNA 链间交联的事实,促使我们研究细胞系模型中的磷酸化蛋白质组 3。CKG 对表达 CT45 的细胞与对照细胞的信号反应的默认分析确实揭示了相关 DNA 损伤通路的激活。
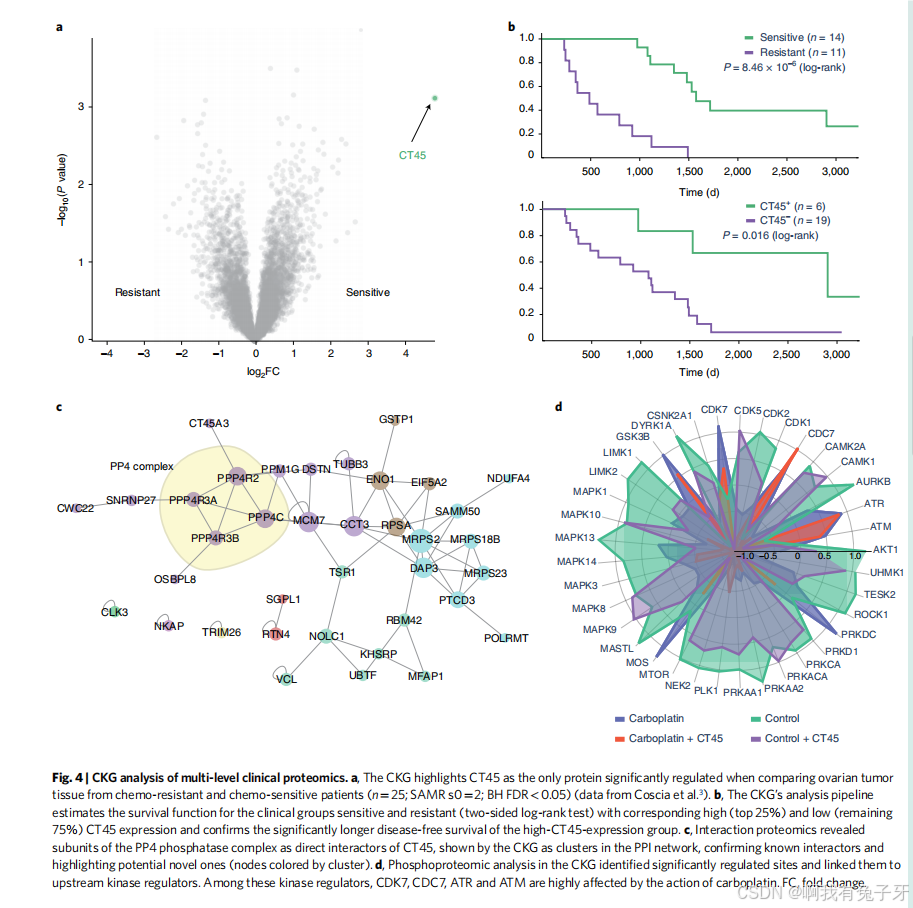
{图 4 | 多层次临床蛋白质组学的 CKG 分析。a,在比较化疗耐药和化疗敏感患者的卵巢肿瘤组织时,CKG 强调 CT45 是唯一显着调节的蛋白质 ( =25 :SAMR s0=2 BHFDR<0.05 (数据来自 Coscia 等人)。b,CKG 的分析管道估计了临床组敏感和耐药组(双侧对数秩检验)的生存函数,相应的高 (前 25%) 和低 (剩余 75%) CT45 表达,并确认高 CT45 表达组的无病生存期显着延长。c,相互作用蛋白质组学揭示了 PP4 磷酸酶复合物的亚基是 CT45 的直接相互作用子,由 CKG 显示为 PPI 网络中的簇,证实了已知的相互作用子并突出了潜在的新相互作用子(由簇着色的节点)。d,CKG 中的磷酸化蛋白质组学分析确定了显着调节的位点,并将它们与上游激酶调节因子联系起来。在这些激酶调节因子中,CDK7、CDC7、ATR 和 ATM 受卡铂作用的高度影响。FC,换折。}
CKG 确定了几种已知的 DNA 损伤激酶 (即 ATM/ATR) 及其相应的调节底物 (SMC1A_ S966/S957、NBN_S615 和 PBRM1_S948),为卡铂的作用机制及其通过 DNA 损伤修复对增殖的负调控提供了更深入的见解。此外,CKG 还揭示了其他几种无法进行人工分析的相关激酶和关联,例如 MAPK 活性的位点特异性激活以及 CDC7 和 CDK7 底物调节的差异(图 4d)。虽然本例中未使用,但 CKG 包括用于基因组和转录组学数据以及其他组学数据类型的类似功能,以便进一步集成。
使用 CKG 确定化疗难治性病例的治疗方案的优先顺序。在用尽标准治疗方案后
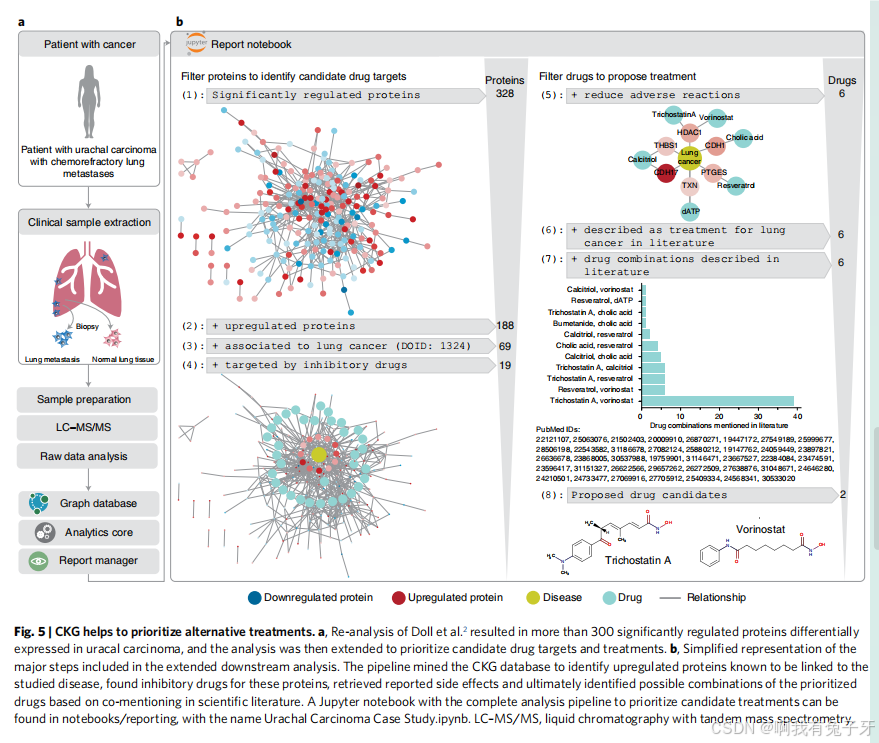
对于终末期癌症,分子分析可能仍会揭示可成药的靶点和药物再利用的机会 51,52,我们之前使用癌症组织的蛋白质组学分析来确定替代的靶向策略 2,4。为了达到类似的目的,CKG 目前挖掘了超过 350,000 个蛋白质与针对它们的已批准或研究药物之间的联系(补充表 4)。
在我们之前对化疗难治性转移性脐尿管癌病例的蛋白质组学研究中,我们提出赖氨酸特异性组蛋白脱甲基酶 1 (LSD1/KDM1A) 作为可能的成药靶点 2。在这里,我们通过扩大的默认分析扩展了该研究,并辅以 Jupyter 笔记本,这些笔记本在可应用于其他研究的可重用管道中根据先验知识实施再利用(图 5)。将肺肿瘤与非癌性组织进行比较,发现数百个显著的
受监管的蛋白质;因此,一种知识衍生的优先排序策略,例如文本挖掘和疾病与药物关联,
成为必要的 51,53,54。CKG 挖掘了图表以确定
文献中共同提及的药物 - 靶标 - 疾病三胞胎(330 万篇文献提及三胞胎); 列举与药物相关的副作用(72,000 个关联); 根据副作用、适应症和靶点寻找类似的药物;并将药物与功能途径联系起来。
在 328 种差异调节蛋白中,188 种上调 (配对 t 检验;补充表 2),其中 69 例已知与肺癌相关。CKG 不仅自动将 LSD1/KDM1A 与反式环丙胺(肿瘤委员会批准用于我们患者的药物)连接起来,而且还表明反式 - 2 - 苯基环丙胺(一种已知的有效去甲基化酶抑制剂)作为另一种治疗选择 55。我们确定了 60 种靶向 19 种优先蛋白质的潜在药物。在检索到与所用化疗方案和确定的抑制剂相关的报告副作用后,CKG 根据不同的副作用(Jaccard 指数)对其余药物进行了重新排序。小于 0.2 的临界值导致 6 种药物 (胆酸、dATP、白藜芦醇、骨化三醇、伏立诺他和曲古抑菌素 A) 靶向 6 种蛋白 (HDAC1、THBS1、CDH1、CDH17、PTGES 和 TXN)。
此外,CKG 建议出版物将这些药物与其蛋白质靶点以及疾病和受影响的组织共同提及,这在 30 多篇出版物中强调了伏立诺他和曲古抑菌素 A 的组合。这些药物抑制 HDAC1,HDAC1 是一种组蛋白脱乙酰酶,可诱导与肿瘤进展相关的表观遗传抑制。此类抑制剂的组合可抑制
表观遗传沉默及其恶性影响 56-58。由
HDAC1 和 LSD1/KDM1A 都参与组蛋白修饰,我们扩展了分析以发现这些蛋白质之间可能存在的联系,这表明它们是 CoREST 复合物的亚基,该复合物最近引起了治疗兴趣,为此 CKG 检索了一篇描述 HDAC1 和 LSD1 抑制的论文(参考文献 59)。
案例研究展示了 CKG 在可共享笔记本中的功能。我们的报告目录(补充图 6)包括重现上述 NAFLD 和尿管癌研究的分析序列,以及对其他四个数据集的重新分析,这些数据集说明了 CKG 在不同情况下的功能。首先,我们重新分析了我们对棕色和白色脂肪细胞之间差异的蛋白质组学研究 60,优先考虑并注释了已知与代谢疾病相关的显着调节蛋白。生成的知识子图
强调了以前我们没有注意到的几个炎症过程和与淀粉样变性的联系(补充图 7)。接下来,CKG 分析了我们小组以外的三项研究,从纵向 COVID-19 Olink 数据集开始,我们在其中重现了比较 COVID-19 阳性和 COVID-19 阴性的结果 61,还调查了队列内严重程度组之间的蛋白质组学差异。该分析强调了 IL-6、IL-17C、CXCL10 和 CCL7 等蛋白质的上调,这些蛋白质通常随着严重程度的增加而增加,并表明 CKG 不仅限于基于 MS 的蛋白质组学数据。髓母细胞瘤多层次蛋白质组学数据集重新分析了串联质量标签蛋白质组学数据集,并采用相似性网络融合来整合蛋白质组学、PTM 和 RNA 测序数据,以揭示髓母细胞瘤亚组以及驱动这些亚组的特征(补充表 5)62。最后,在我们对 CPTAC 胶质母细胞瘤发现研究 (https://cptac-data-portal.georgetown.edu/study-summary/S048) 的再分析中,我们使用知识图谱来探索可能的药物抑制剂,以比较肿瘤和正常脑组织时发现的显着上调的蛋白质列表(补充表 6)。这项探索性分析将肿瘤中许多上调的蛋白质与吡非尼酮联系起来,吡非尼酮是一种通常用于特发性肺纤维化的药物,已知可抑制 TGF-β 信号传导,TGF- 信号传导是在 malig 中失调的途径
NANT 神经胶质瘤,并且还减少了肿瘤细胞外基质 63,64。
所有这些笔记本都是 CKG 代码存储库和文档的一部分,可以在 https://CKG.readthedocs.io/en/ latest/advanced_features/ckg-notebooks.html 上可视化。
讨论
CKG 代表大型网络中的先验知识、实验数据和去标识化的临床患者信息。它使用图形结构将蛋白质组学数据与所有这些信息协调一致,该图形结构自然地提供了与已鉴定蛋白质的直接联系。我们发现它的自动化、即时和迭代性质有助于揭示相关的生物学背景,以便更好地理解和产生新的假设。此外,图形结构提供了一个灵活的数据模型,可以轻松扩展到新的节点和关系。尽管 CKG 是专门为回答临床相关问题而设计的,但它同样适用于其他生物体和任何生物学研究 65。
CKG 的分析核心采用完全用 Python 实现的开放式模块化设计,利用广泛使用和维护良好的开源库,这些库涵盖了广泛的数据科学生态系统:统计、网络分析、机器学习和可视化。使用这些库可确保底层算法和方法的质量、稳健性和效率。
它还支持整合数据科学的新发展,可以快速适应以专门支持蛋白质组学数据分析。CKG 还解决了对科学结果可重复性的日益担忧 66,67。我们使用 Jupyter Notebook 来生成可共享的分析管道,使结果具有可重复性和可复制性,并设想蛋白质组学界及其他领域广泛采用该框架。
为了促进 CKG 的采用,我们集成了社区制定的标准,例如 mzTab 和样本和数据关系格式 (SDRF)68,用于元数据,以及由商业或开源软件生成的多种蛋白质组学数据格式。此外,CKG 的模块化设计有助于更改这些格式和合并新的格式。这种灵活性确保 CKG 能够跟踪不断扩大和活跃的基于 MS 的蛋白质组学社区以及其他组学计划。
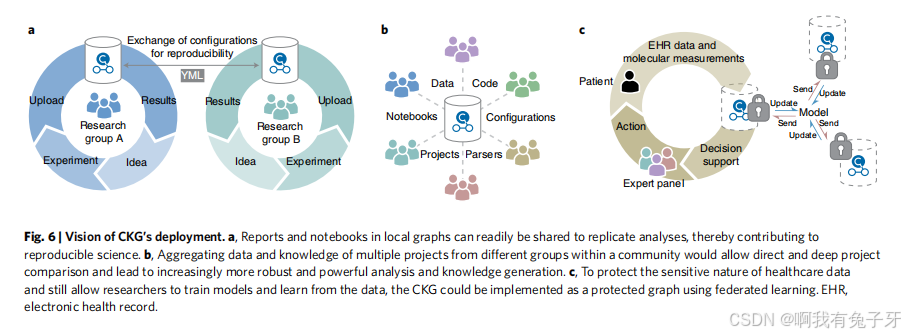
CKG 的不同组件允许各个研究小组分析、整合和构建其蛋白质组学和其他组学项目的数据库。可以轻松共享报告和笔记本以复制分析,从而有助于可重复的科学(图 6a)。除此之外,CKG 的开放性和免费可用性可能允许将数据和知识聚合在我们所说的社区图谱中(图 6b)。这将确保社区从其他地方进行的类似蛋白质组学或组学项目中受益。对于生物标志物的发现,这构成了 “矩形策略” 69 的延伸,允许直接和深入的项目比较,并导致越来越强大和强大的分析和知识生成。
我们设想不同的团体和机构将拥有自己的本地版本的 CKG,以保护医疗保健数据的敏感性,但仍然支持跨平台分析。新方法,例如差分隐私和联合
learning70,71 的 c. 将允许研究人员使用 CKG 来训练模型
在没有直接访问敏感数据的机构之间迭代(图 6c)。人工智能将在基于 MS 的蛋白质组学和生物标志物发现中发挥越来越大的作用 72,我们期待将 CKG 与这些功能集成,并利用新的图深度学习功能。
总之,我们描述了 CKG,这是一个开放、强大的框架,用于蛋白质组学和多层次组学数据的透明、自动化和综合分析,旨在结合可重复科学的所有先决条件。因此,CKG 直接解决了个性化医疗和严格的、数据驱动的临床决策过程的一些主要瓶颈。我们预计生物医学和临床研究界的其他人将受到鼓励,为这个平台做出贡献并进一步发展。
在线内容
任何方法、其他参考文献、Nature Research 报告摘要、源数据、扩展数据、补充信息、致谢、同行评审信息;作者贡献和利益争夺的详细信息;数据和代码可用性声明可在 Production and detection of cold antihydrogen atoms | Nature S41587-021-01145-6 上获得。
收稿日期: 2020-11-18; 录用日期: 2021-11-1; 在线发布:2022 年 1 月 31 日


















 529
529

 被折叠的 条评论
为什么被折叠?
被折叠的 条评论
为什么被折叠?
 到【灌水乐园】发言
到【灌水乐园】发言









