







1. NanoVar简介
NanoVar 是一种新颖的 SV 识别工具,在准确性和速度方面表现出色,同时克服了三代测序全基因组测序(WGS)低深度和易出错的问题 。在模拟数据中, NanoVar 在仅使用 4 - 8X 覆盖度的数据集检测纯合和杂合 SV ,优于现有的 三代测序结构变异(SV)检测工具,实现了较高的 SV 检测准确率(F1>0.92)。NanoVar 的性能在低深度真实数据(如样本 NA12878)和临床数据中也得到了体现,并可靠地推断 SV 的类别、大小和杂合性。模拟和真实数据的结果都表明,4X 测序深度可能不足以全面发现杂合 SV,建议使用 8X 或更高测序深度。
参考文献:
Tham, CY., Tirado-Magallanes, R., Goh, Y. et al. NanoVar: accurate characterization of patients’ genomic structural variants using low-depth nanopore sequencing. Genome Biol. 21, 56 (2020). https://doi.org/10.1186/s13059-020-01968-7
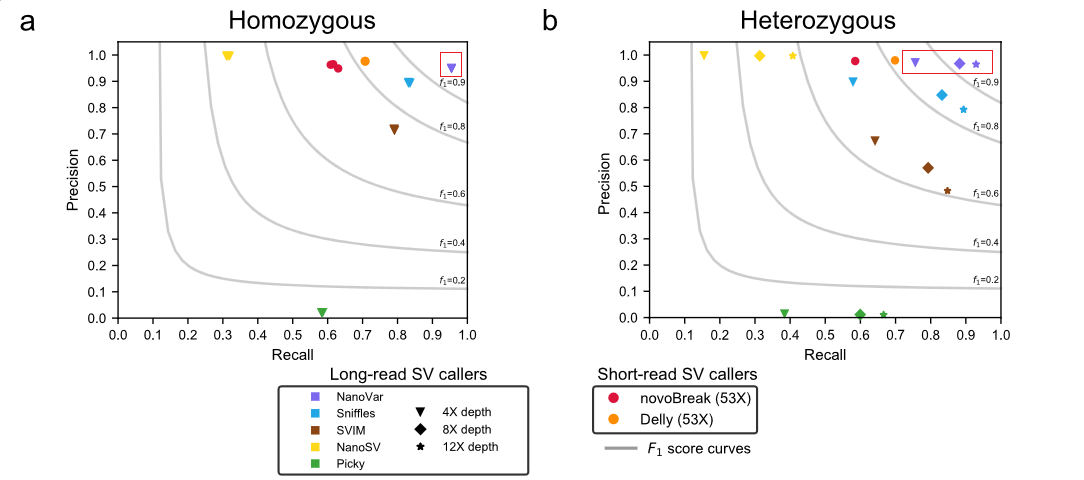
使用NA12878标准品,在4X深度下,不同的三代SVs检测工具,检测缺失SVs时SVIM召回率最高(0.78)、nanoVar (0.71)、 Sniffles (0.58),and Picky(0.54); 对于插入SVs,nanovar召回率最高(0.66))、SVIM (0.62),、Sniffles (0.28) and Picky (0.22)。在8X深度下,nanova和SVIM召回率接近0.82。
github地址: https://github.com/benoukraflab/NanoVar
不同测序深度(4X 8X 12X)纯合/杂合SVs检测准确率和召回率:
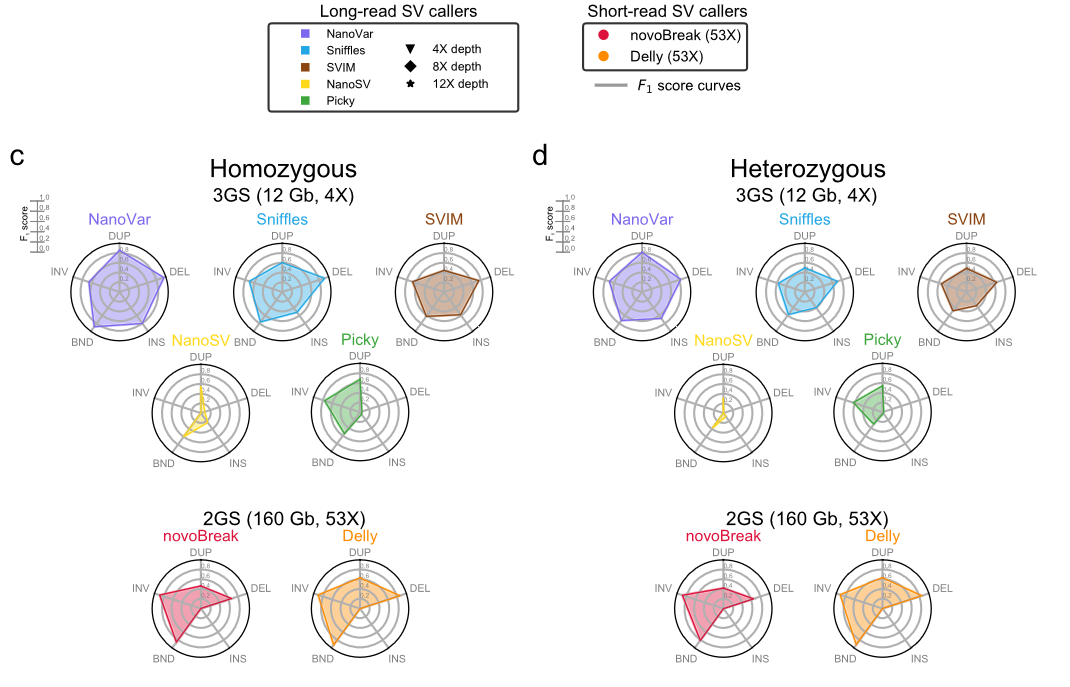
2代测序和3代测序SVs检测工具比较:
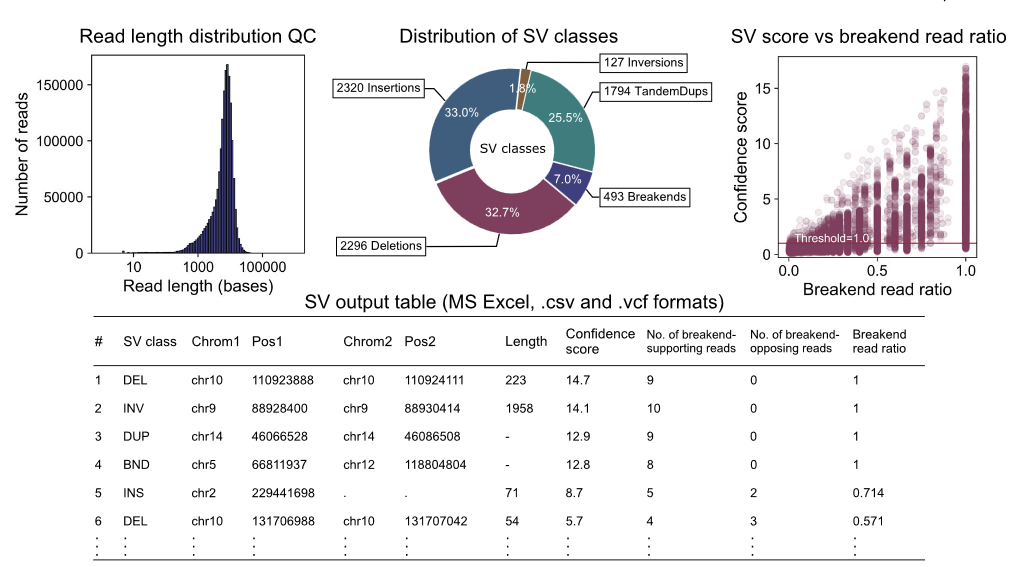
输出结果:
2. 安装依赖
软件版本要求:
- bedtools >=2.26.0
- samtools >=1.3.0
- minimap2 >=2.17
conda create -n nanovar -c bioconda python=3.9
conda activate nanovar
conda insatall bedtools -y
conda install samtools==1.3.0 -y
conda install minimap2==2.17 -y
conda install seqtk -y
# 安装nanovar
conda install nanovar==1.8.0 -y
3. 基本用法
# -t: 线程数
# -x: 三代测序数据类型,支持ont, pacbio-clr, pacbio-ccs
# -f: 参考基因组gap bed文件或调用内置hg19、hg38和mm10 gap bed
# sample.bam: 样本碧迪排序后BAM文件
# ref.fa: 参考基因组fasta文件
# working_dir: 工作目录
# ONT数据
nanovar -t 10 -x ont -f hg38 sample.bam ref.fa working_dir
4. 参数说明
usage: nanovar [options] [FASTQ/FASTA/BAM/CRAM] [REFERENCE_GENOME] [WORK_DIRECTORY]
positional arguments:
[FASTQ/FASTA/BAM/CRAM]
Path to long reads or mapped BAM/CRAM file.
Formats: fasta/fa/fa.gzip/fa.gz/fastq/fq/fq.gzip/fq.gz/bam/cram
[reference_genome] Path to reference genome in FASTA. Genome indexes created
will overwrite indexes created by other aligners such as bwa.
[work_directory] Path to work directory. Directory will be created
if it does not exist.
options:
-h, --help show this help message and exit
-x str, --data_type str
Type of long-read data [ont]
ont - Oxford Nanopore Technologies
pacbio-clr - Pacific Biosciences CLR
pacbio-ccs - Pacific Biosciences CCS
-f file, --filter_bed file
BED file with genomic regions to be excluded [None]
(e.g. telomeres and centromeres) Either specify name of in-built
reference genome filter (i.e. hg38, hg19, mm10) or provide full
path to own BED file.
--annotate_ins str Enable annotation of INS with NanoINSight,
please specify species of sample [None]
Currently supported species are:
'human', 'mouse', and 'rattus'.
-c int, --mincov int Minimum number of reads required to call a breakend [4]
-l int, --minlen int Minimum length of SV to be detected [25]
-p float, --splitpct float
Minimum percentage of unmapped bases within a long read
to be considered as a split-read. 0.05<=p<=0.50 [0.05]
-a int, --minalign int
Minimum alignment length for single alignment reads [200]
-b int, --buffer int Nucleotide length buffer for SV breakend clustering [50]
-s float, --score float
Score threshold for defining PASS/FAIL SVs in VCF [1.0]
Default score 1.0 was estimated from simulated analysis.
--homo float Lower limit of a breakend read ratio to classify a homozygous state [0.75]
(i.e. Any breakend with homo<=ratio<=1.00 is classified as homozygous)
--hetero float Lower limit of a breakend read ratio to classify a heterozygous state [0.35]
(i.e. Any breakend with hetero<=ratio<homo is classified as heterozygous)
--sv_bam_out Outputs a BAM file containing only SV-supporting reads with
their corresponding SV-ID(s) stored in the "nv" tag separated by comma.
--debug Run in debug mode
-v, --version Show version and exit
-q, --quiet Hide verbose
-t int, --threads int
Number of available threads for use [1]
--model path Specify path to custom-built model
--mm path Specify path to 'minimap2' executable
--st path Specify path to 'samtools' executable
--ma path Specify path to 'mafft' executable for NanoINSight
--rm path Specify path to 'RepeatMasker' executable for NanoINSight
生信软件文章推荐
生信软件1 - 测序下机文件比对结果可视化工具 visNano
生信软件3 - mapping比对bam文件质量评估工具 qualimap
生信软件4 - 拷贝数变异CNV分析软件 WisecondorX
生信软件7 - 多线程并行运行Linux效率工具Parallel
生信软件8 - bedtools进行窗口划分、窗口GC含量、窗口测序深度和窗口SNP统计
生信软件9 - 多公共数据库数据下载软件Kingfisher
生信软件10 - DNA/RNA/蛋白多序列比对图R包ggmsa
生信软件11 - 基于ACMG的CNV注释工具ClassifyCNV
生信软件12 - 基于Symbol和ENTREZID查询基因注释的R包(easyConvert )
生信软件13 - 基于sambamba 窗口reads计数和平均覆盖度统计
生信软件14 - bcftools提取和注释VCF文件关键信息
生信软件15 - 生信NGS数据分析强大的工具集ngs-bits
生信软件17 - 基于fasta文件的捕获探针设计工具catch
生信软件18 - 基于docker部署Web版 Visual Studio Code
生信软件20 - seqkit+awk+sed+grep高级用法技巧合辑
生信软件21 - 多线程拆分NCBI-SRA文件工具pfastq-dump
生信软件22 - 测序数据5‘和3‘端reads修剪工具sickle
生信软件23 - Samtools和GATK去除PCR重复方法汇总
生信软件24 - 查询物种分类学信息和下载基因组TaxonKit和ncbi-genome-download
生信软件26 - BWA-MEM比对算法性能更好的bwa-mem2
生信软件27 - 基于python的基因注释数据查询/检索库mygene
生信软件28 - fastq与bam的reads数量计算与双端fastq配对检测工具fastq-pair
生信软件29 - 三代数据高效映射精确的长读段比对工具mapquik
生信软件31 - Bcftools操作VCF/BCF文件高级用法合集
生信软件33 - Wgsim生成双端(PE) fastq模拟数据
生信软件34 - 大幅提升Python程序执行效率的工具Pypy
生信软件36 - SAM/BAM/CRAM文件插入SNV/INDEL/SV工具Bamsurgeon
生信软件37 - 基于测序reads变异进行单倍型分型工具WhatsHap
生信软件39 - GATK最佳实践流程重构,提高17倍分析速度的LUSH流程
生信软件43 - iGenomes批量下载Ensembl、NCBI、GATK和UCSC参考基因组和注释文件资源



















 被折叠的 条评论
为什么被折叠?
被折叠的 条评论
为什么被折叠?
 到【灌水乐园】发言
到【灌水乐园】发言











