







1、前言
这个问题正如黄树嘉老师知乎文章中所说,向左移、向右移都是可以的,但要在一定的文件格式中下定义,他文章提到的是vcf。
描述一个突变有多种文件, bam => vcf = > 注释文件 => 结果文件。
2、在bam文件中

上面图片来自黄树嘉老师知乎文章,可以看出都是缺失了 “GC” 碱基,从比对角度看,结果一样。可以分别认为它们在左、中、右发生的,都没问题,这时候就需要比对软件有约定俗称的规则,目前是左移。
通常情况下BWA文件的indel突变位置都是左移的。如果发现不是这样,想要对bam文件进行左移,可以使用GATK软件自带的矫正功能,不同的版本模块名称是不同的,需要现查。
3、在vcf文件中
通常情况下拷变异的主流软件都是 变异 向左 “贪婪比对” 的,也就是说会 一直向左比对,直到不匹配为止,然后以最左边的碱基位置表示变异的起始位置。
变异表示的标准化包含两个部分:
1,节俭原则 2,左对齐。
这两部分分别对应变异的长度与位置。
可以查看你手里的vcf、与别人给你的vcf关于indel位点是左移还是右移的。依据实际情况进行软件矫正(vt、bedtools,使用前请验证),或者自己写脚本实现。
4、在注释文件中
此时要用注释软件对vcf文件进行注释,增加突变的基因、C点、P点等信息。按照目前业内共识、临检中心室间质评要求,都要求注释结果要符合 HGVS规范。
因为临床医生看突变更考虑功能影响,所以HGVS规范更贴合临床医生的要求。
HGVS命名规范,是人类基因组变异协会制定的对突变进行命名的规范(sequence variant nomenclature by human genome variation society),即当序列改变和氨基酸功能一致等价时,突变的命名要以靠近 3-UTR 区来命名。

概括下就是 “ 3’ 规则 ”:所有变异都应按照靠近基因转录方向末端(3’端)的位置来表述。所以在实际注释的时候,要根据基因所在strand来确定indel的注释方向。
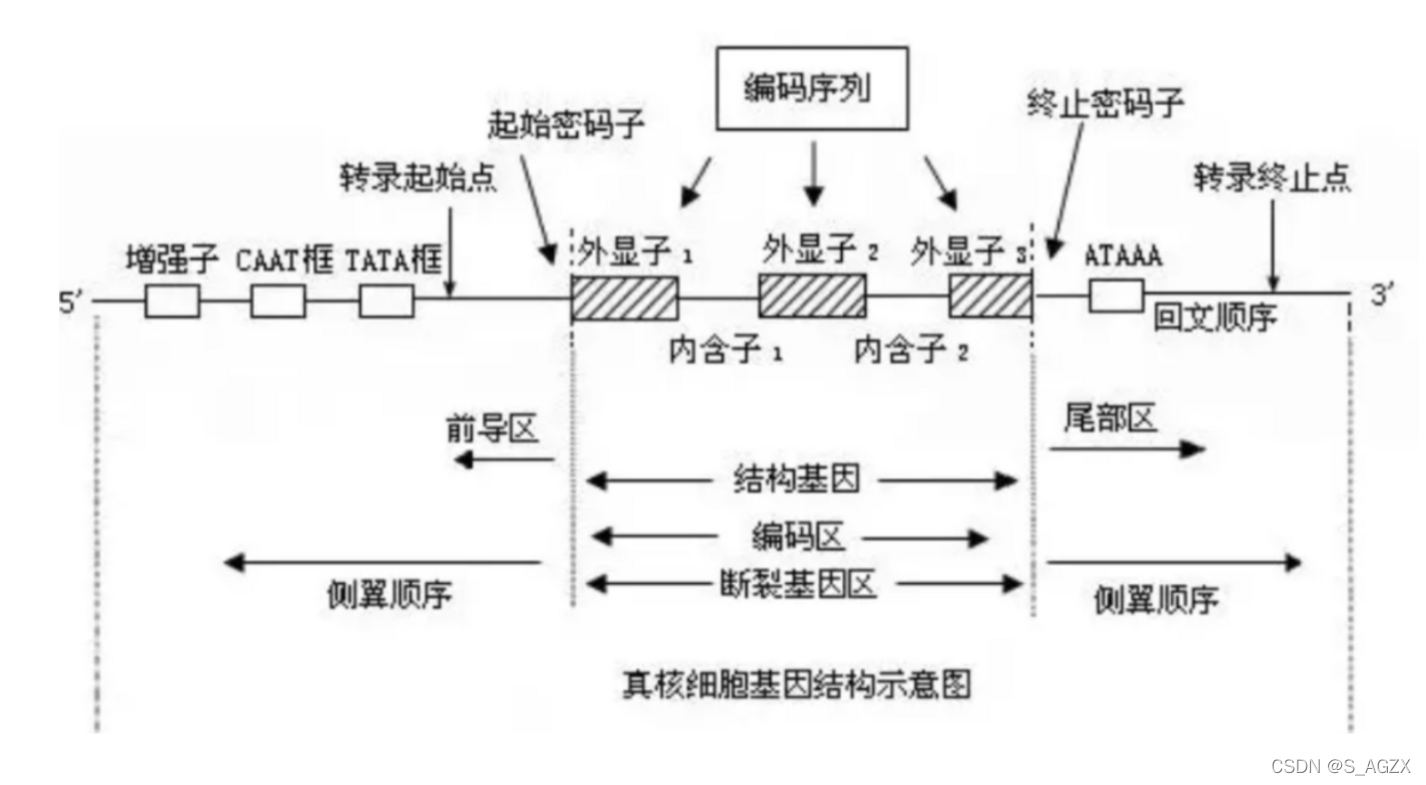
hg19文件chr1 -> X染色体,1 -> 20000000+位点的过程中,不同基因在参考序列上是不同的strand。转录的时候,负链基因的3’端在左侧,正链基因的3’端在右侧,于是依据3’ 规则,负链基因的突变应该向左贪婪比对,正链基因的位点向右贪婪比对(“正链基因右对齐,负链基因左对齐”)。目前的主流注释软件VEP、ANNOVAR都不能直接给出完美符合“HGVS规范”的注释结构。
5、在结果文件中
通常结果文件直接从注释文件中获取,此处不再说明。
参考文件:
1、关于Indel变异,我该往左还是该往右?
2、Variant Normalization 变异的标准化
3、Unified representation of genetic variants
4、人类全基因组遗传变异解读的高通量测序数据规范

















 1118
1118

 被折叠的 条评论
为什么被折叠?
被折叠的 条评论
为什么被折叠?
 到【灌水乐园】发言
到【灌水乐园】发言









