






欢迎关注微信公众号(医学生物信息学),医学生的生信笔记,记录学习过程。
安装及加载R包
BiocManager::install("sva")
BiocManager::install("bladderbatch")
library(sva)
library(bladderbatch)
ComBat函数
加载示例数据
data(bladderdata)
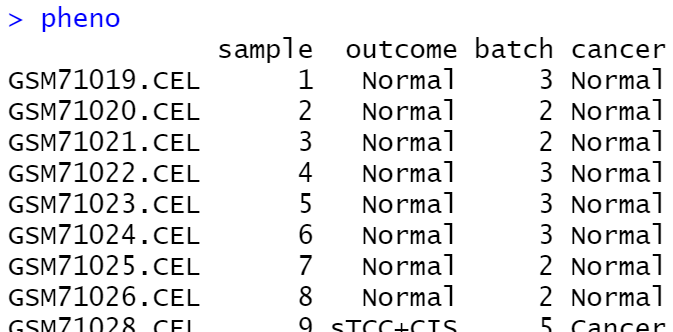
pheno = pData(bladderEset)
edata = exprs(bladderEset)
edata[1:6,1:6]
# edata=log2(edata+1)


提取批次信息
# 注意:批次信息的顺序应该与数据框中样本的顺序相匹配
batch = pheno$batch
# 根据自己的项目可自定义批次,可用下方代码
# batch <- rep(c("n1","n2"),each = 20 )
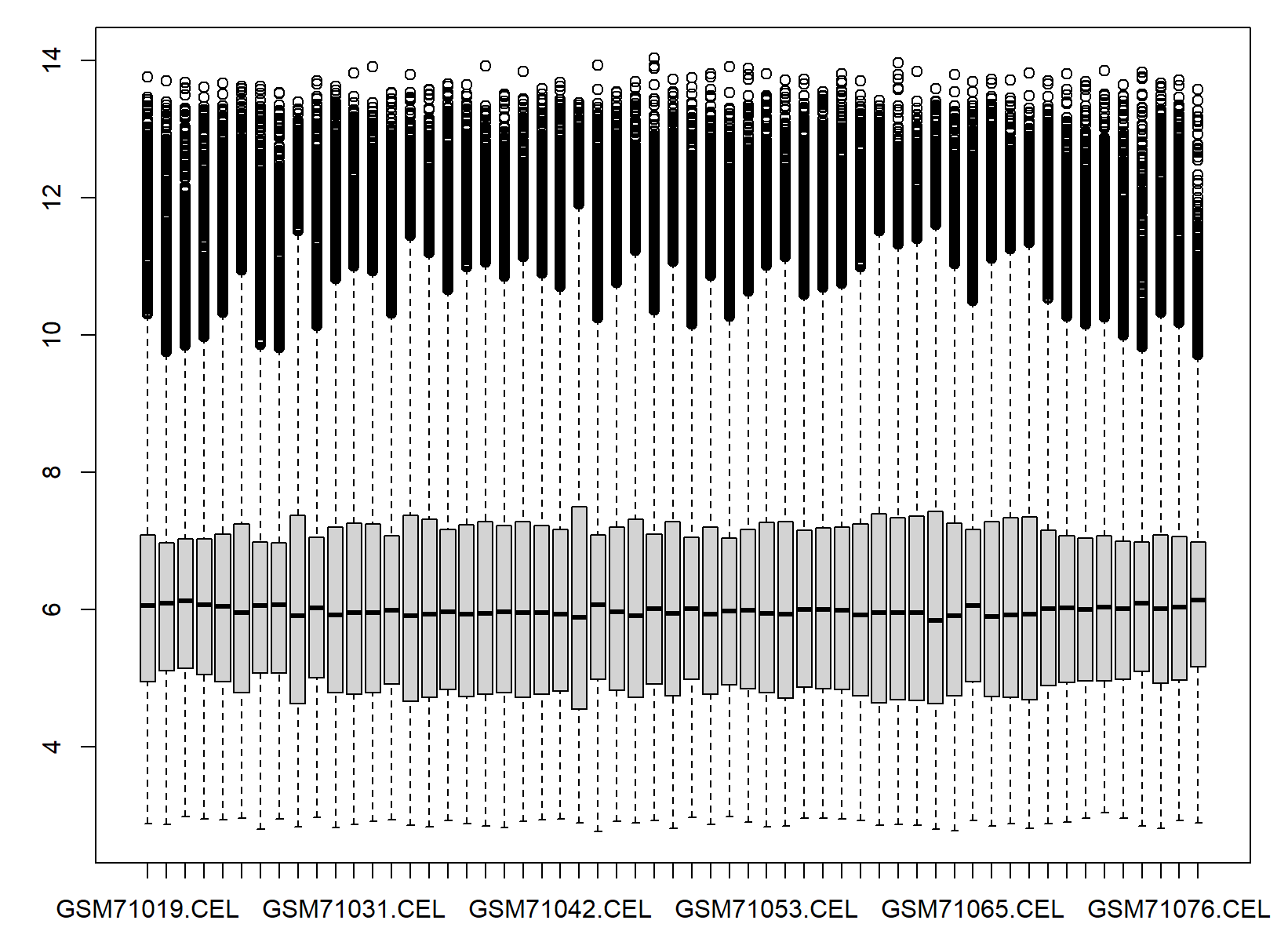
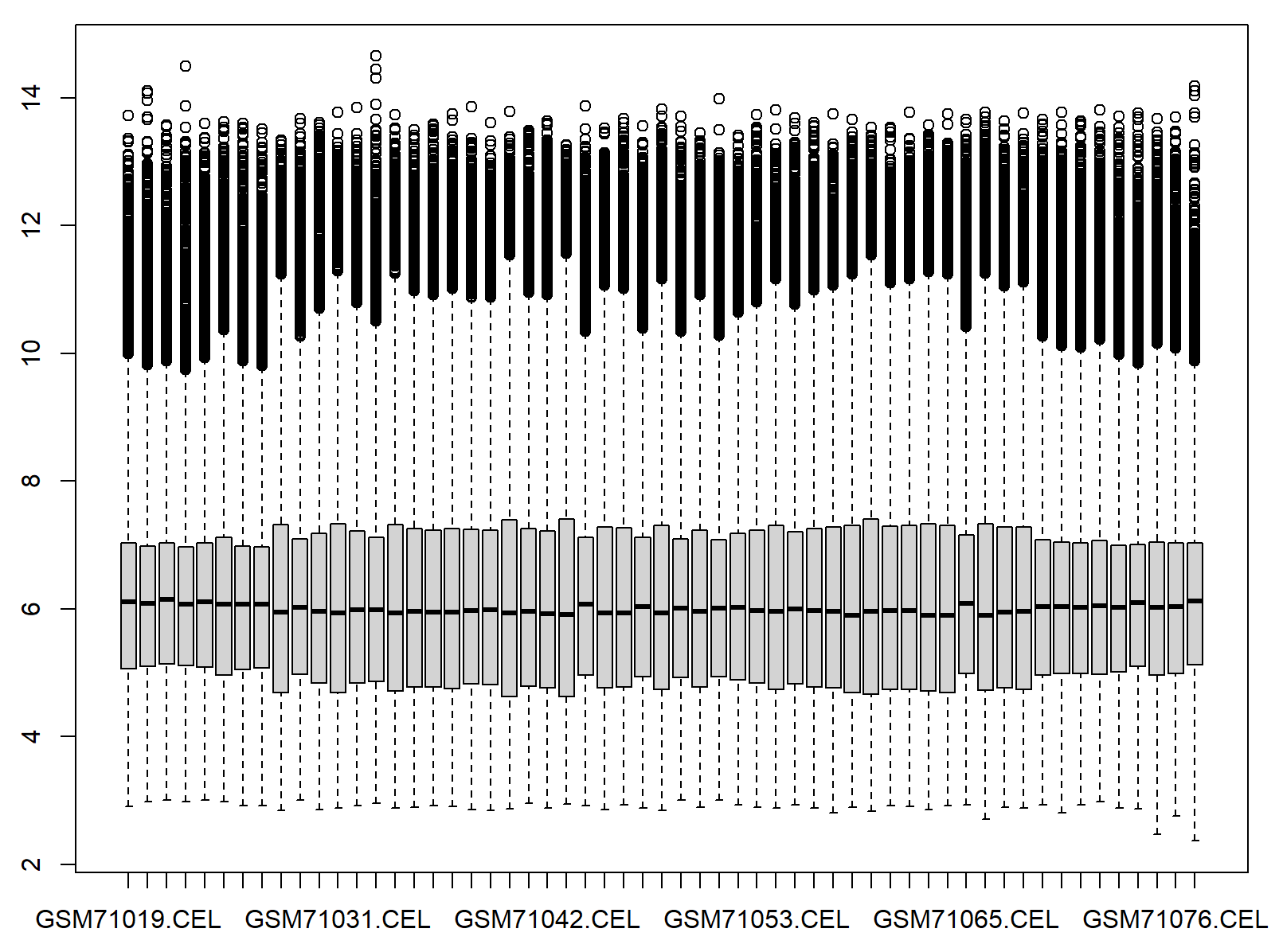
查看未批次矫正的数据分布
boxplot(edata)
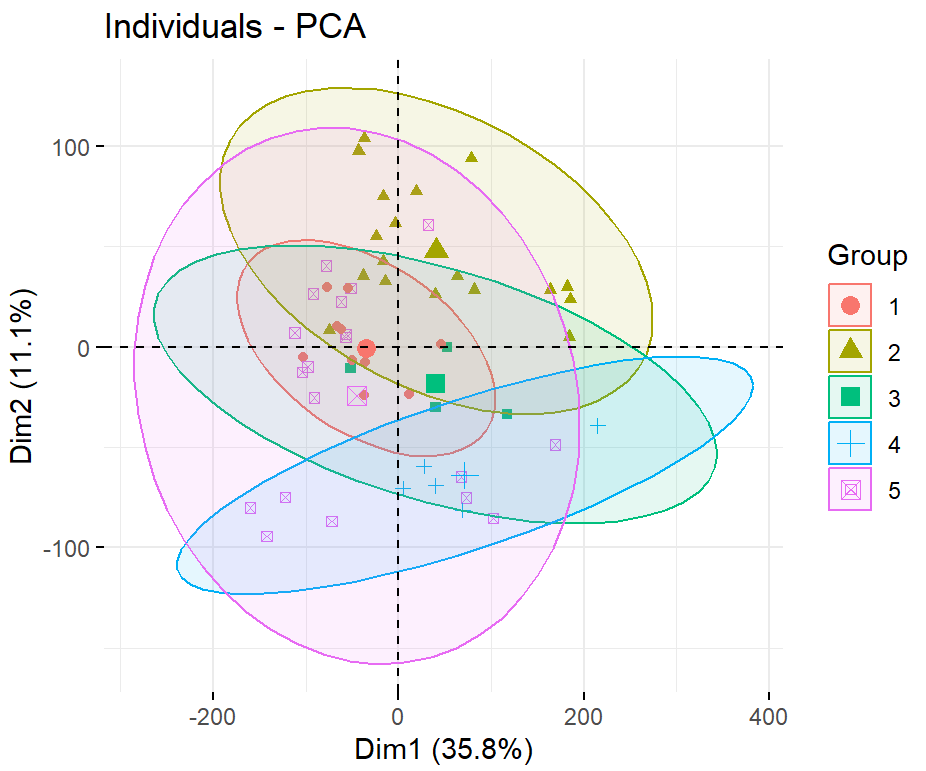
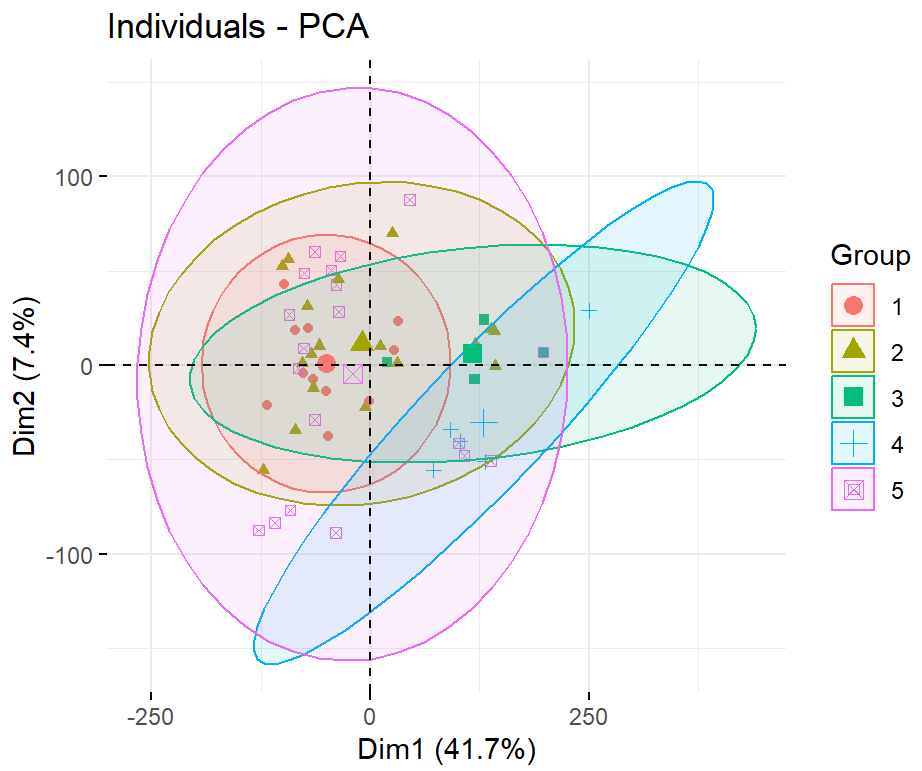
library(FactoMineR)
library(factoextra)
pre.pca <- PCA(t(edata),graph = FALSE)
fviz_pca_ind(pre.pca,
geom= "point",
col.ind = as.factor(batch),
addEllipses = TRUE,
legend.title="Group")
批次矫正
对已知的批次进行批次矫正。
combat_edata = ComBat(dat=edata, batch=batch, mod=NULL, par.prior=TRUE, prior.plots=F)
combat_edata[1:6,1:6]
save(combat_edata,pheno,file = "RNA_seq.rdata")
批次矫正后的数据:

指定已有分组,防止过度矫正
进行批次矫正时,为了防止过度矫正,从而掩盖了原本的生物学差异。我们可以通过ComBat函数的mod参数来指定实验处理的分组,保留生物学差异。这里选取pheno$cancer。
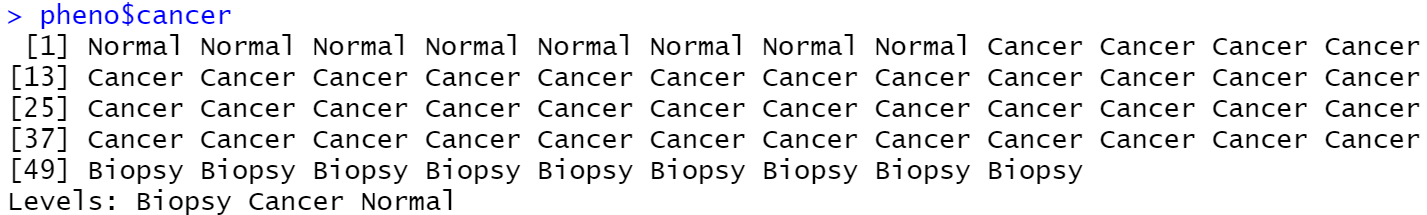
mod <- model.matrix(~factor(pheno$cancer))
# mod = model.matrix(~as.factor(cancer), data=pheno)
combat_edata = ComBat(dat=edata, batch=batch, mod=mod, par.prior=TRUE, prior.plots=F)
combat_edata[1:6,1:6]

查看批次矫正后的数据分布
boxplot(combat_edata)
library(FactoMineR)
library(factoextra)
pre.pca <- PCA(t(combat_edata),graph = FALSE)
fviz_pca_ind(pre.pca,
geom= "point",
col.ind = as.factor(batch),
addEllipses = TRUE,
legend.title="Group")
ComBat-Seq
可以通过sva包中ComBat-Seq函数来对RNA-Seq count型数据去批次。
示例数据
count_matrix <- matrix(rnbinom(400, size=10, prob=0.1),
nrow=50, ncol=8)
batch <- c(rep(1, 4), rep(2, 4))

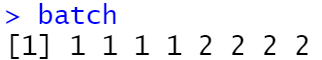
批次矫正
adjusted <- ComBat_seq(count_matrix, batch=batch, group=NULL)

保留单个生物学分组的批次矫正
加入生物学分组信息,在批次矫正时,保留生物学差异,防止原本的生物学差异被矫正。
# 自定义分组信息
group <- rep(c(0,1), 4)

adjusted_counts <- ComBat_seq(count_matrix, batch=batch,
group=group)
保留多个生物学分组的批次矫正
# 自定义分组信息
cov1 <- rep(c(0,1), 4)
cov2 <- c(0,0,1,1,0,0,1,1)
covar_mat <- cbind(cov1, cov2)

adjusted_counts <- ComBat_seq(count_matrix, batch=batch,
group=NULL, covar_mod=covar_mat)















 4852
4852











 被折叠的 条评论
为什么被折叠?
被折叠的 条评论
为什么被折叠?
 到【灌水乐园】发言
到【灌水乐园】发言









