








最近在进行单细胞相关的数据的处理,由于样本是不同的病理时期的,料想它们在测序的时候也是不同步进行的,因此认为批次效应也肯定是存在的,但是之前没有解除过这方面的问题,在网上看了很多技术帖,有一点基础的了解,再进一步的看看文献原著,希望深入的了解单细胞的批次效应,以及在不同的样本数据情况下,用何种批次处理效果好,以及批次处理效果的标准如何评定。
 A benchmark of batch-effect correction
A benchmark of batch-effect correction
methods for single-cell RNA sequencing
data
用于普通的RNAseq数据整合时的批次矫正分析,如ComBat 和limma,其也可以用于单细胞批次矫正处理。但是由于单细胞数据的drop out以及RNA 扩增和捕获过程中的失败,因此需要新的批次处理的技术。
然而近期由Haghverdi提出的一个流行,且受欢迎的方法MNN(mutual nearest neighbors)可用于2个数据集之间的分析,在2个数据集中,配对的细胞列表用于计算翻译向量,进而在共享空间比对。这种方法的优点是数据进行了归一化处理,因而可用于下游的分析。然而这种方法需要在高维基因表达空间计算临近细胞列表,因而费时费力。需要进一步的更进。
基于上述的MNN 方法,研究者进一步的开发了新的算法即FastMNN,这种方法通过使用PCA分析在亚空间里使用MNN的方法,进而在运行时间和精确性上进一步得到提升。
其他的2种方法,Scanorama and BBKNN也通过寻找亚空间的方法来运用MNN。
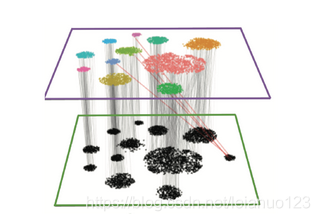
对于我们最常见的单细胞分析的软件Seurat来说,它在 进行细胞的批次处理和数据整合的时候,进行了更新换代,最开始的版本2017年,Satjia lab开发了Multi canonical correlation analysis (CCA)的算法来对数据进行降维,然后再最相关的数据特征,然后进行批次数据的比对。在Seurat3.0以上的版本中即Seurat Intergation,其对批次矫正的算法进行了更新,主要是2步,首先通过CCA 的算法对数据进行降维得到亚空间,然后进一步的通过 MNN的算法对CCA亚空间的细胞进行计算,MNN的算法类似于一个anchors 的作用,将2个数据集的细胞进行锚定,如图。
除了上述提到的方法,近期还提到了其他的方法,比如说Harmony,其首先利用PCA进行降维,在低维PCA的空间里,通过迭代消除当前的批次效应,在每次迭代的过程中,他将不同批次的相似细胞聚类,最大化每个聚类批次的多样性,然后计算应用于每个细胞的批次矫正因子,这种方法能够很快和准确的进行检测。
LIGER是一种新的算法,主要是用于预知其他方法的缺点,它假设每个数据集之间的差异完全是由技术误差导致的,而非生物学效应,那么这种假设将让我们考虑完全除去这些技术误差。LIGER通过使用非负矩阵分解首先获取输入数据的低维表示,该表示法由2部分组成,一组特定批次因子和一组共有因子。此后执行聚类,然后使用共享因子邻域图搜索具有相似邻域的细胞,在已经鉴定的细胞簇,然后将因子加载分位数归一化去匹配选定的参考数据集,从而完成矫正。
机器学习领域深度神经网络近期也被用于单细胞的批次矫正过程。
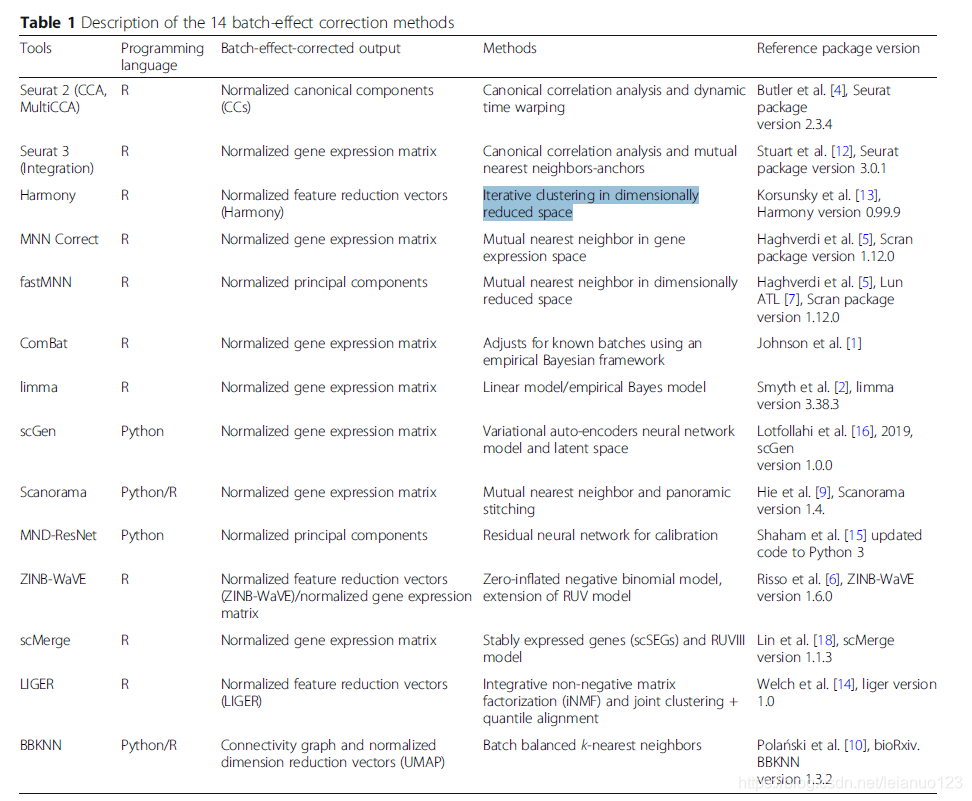
在这篇文章中,作者对以下14个算法MNN,Fast MNN以及MultiCCASeurat 和Seurat3以及MMD-ResNet,Harmony和Scanorama 以及BBKNN和scGen Combat 和LIGER,limma 和scMerge 以及ZINB-WaVE等算法进行了测试,在五种不同的场景下测试了十个数据集,五种场景分别是不同测序技术批次的已知细胞类型;含有未知细胞的批次;多批次;超过百万细胞的大数据;用于基因差异表达的数据集模拟。
结果
通过5项指标来评估14中批次矫正的方法对十个数据集处理的效果
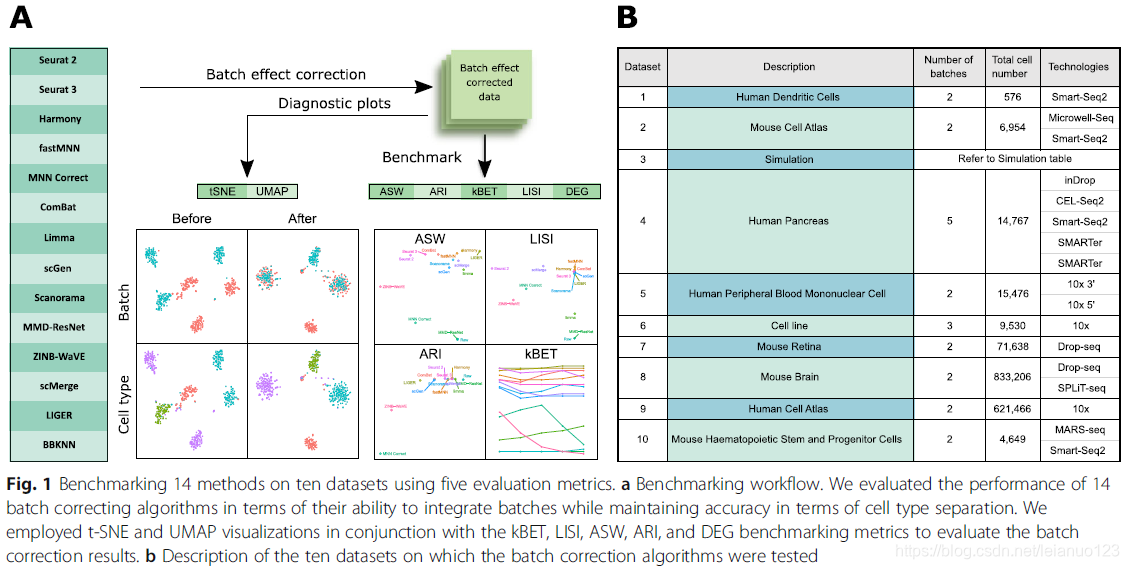 本文中用于批次效应矫正效果评估的五大指标,
本文中用于批次效应矫正效果评估的五大指标,
1.KBET ( k-Nearest neighbor batch-effect test)主要用于评估批次矫正后,不同的批次之间在局部的混合效果,本方法是基于奇异值降解的基础上进行降维。当拒绝率低的时候,表明不同批次混合的结果好(其实他的拒绝率是一个零和假设,也即所有的批次都能够很好的混合,局部分布和整体分布的相似足够大,而不被拒绝说批次混合不好),整个的拒绝率是0-1之间,当拒绝率接近0的时候,就意味着不同批次的细胞批次效应处理的好,细胞能够很好的混合。在这种kbet 的评估中呢,主要是通过批次矫正后的产生PC成分或者是批次降维后的表达矩阵,选取Top 的PC成分,由于K临近值的数目对于对KBET 有很大的影响,因此作者运行KBET通过K值预测的方法,根据KBET文章中的方法,作者选取K输入值分别为样本数量的5%,10%和15%和20%以及25%,运行KBET获得所有的KBET 的拒绝值,然后取中位数,作为最终的KBET值,用于对每种方法进行评估。 最后通过Wilcoxon statistical test和BH 的矫正方法来确认整合输出的结果相比于其他的方法具有显著的统计学意义。
2.LISCI(Local inverse Simpson’s index) 局部辛普森指数 ,也是用来评估局部的水平,来评估细胞的批次和细胞类型混合的结果。其包含iLISI(即整合局部辛普森指数)和相比于KBET固定的K值,LISI主要通过具有固定的复杂度的局部距离分布选择临近的邻居。临近值的选择然后用于计算辛普森指数多样性,这是这个邻域呈现出来的有效类型数,指数通过用来计算批次标签,接近于期待批次数的分数,表明批次混合效果好。其中iLISI分数只计算所有批次中都出现的细胞,对于细胞类型的LISI,这些指数用于计算所有细胞类型的标签,分数接近1表明这种类型的细胞纯度非常高。在本文中作者为每个细胞计算了它们的cLISI值和iLISI值,然后进一步的计算它们的中位数,接着通过扫描最大和最小的分数对中位数进行scale,为了进一步的联合分析细胞的纯度和批次的混合效应,作者通过计算cLISI 和iLISI的调和平均数计算得到F值,高的F值表明批次效应矫正的越好,Wilcoxon统计学检验和Benjamini Hochberg 矫正用于对iLISI 和cLISI的结果进行统计学显著性检测。
调和平均数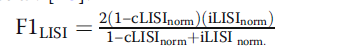
3.ASW (Average silhouette width) 平均轮廓宽度
也是通过联合细胞类型和批次标签来评估批次矫正的效果,数据点的轮廓分数,通过它距相邻的Cluster的平均距离减去其与相同cluster 中的其他成员的平均距离,然后再除以2个值中的较大者,这种结果的值在-1到1之间,值越大表明其在当前的Cluster 中矫正的越好,值越低表明矫正的效果越差。同一个label 内的所有数据点的平均值用来衡量细胞的类型和批次混合的效果。在作者的这项研究中,作者首先将数据集随机抽样到原始数据集的80%,然后对通过向下取样的方法获得的数据集计算其前20个PC作为输入数据来计算数据之间的距离以获得ASW得分,为了确保这个ASW得分的稳定性,作者重复此过程20次,为细胞类型混合和批次效应混合获取20个ASW,然后将批次和细胞类型的中位数用于后续的进一步的计算,作者逆转了批次ASW值(越高越好),然后进一步的将2个ASE值进行归一化到0和1之间,为了进一步的联合分析细胞类型纯度和批次混合效应,作者也进一步的计算了批次和细胞类型SAW值的调和平均数,得到F1值,F1值越大(批次的ASW值越小,细胞类型的ASW值越高)表明批次矫正的效果越好,Wilcoxon统计学检验和Benjamini Hochberg 矫正用于对 ASW结果的检验,来分析此种方法的ASW是否在统计学上优于其他的方法。
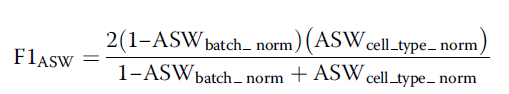
4.ARI (Adjusted rand index)能够通过细胞类型和批次混合结果评估批次矫正的结果,ARI用于评估两个标记列表里的匹配细胞的百分比,在作者的工作中,作者对数据集进行二次取样,取原始数据的80%,然后对二次取样后矫正过的数据获取前20 的PC成分,通过使用K值函数进行K值聚类,K是每个特有的细胞类型数目,为了使用ARI评估每个细胞类型的纯度细胞类型标记的结果与adjustedRandIndex 函数聚类的结果进行比较,高的ARI的结果对应的是高纯度细胞类型,为了批次混合评估,只有出现在所有细胞簇的细胞用于后续的评估,批次标记和K值聚类的结果标记进行比较,低的ARI结果表明好的混合效果。和轮廓系数相似,为了产生稳定的ARI结果,作者重复了20次随机二次取样,为每个i细胞类型和批次获得20个ARI分值,随后将ARI的中位数值进行归一化到0和1,通过调和平均数来获取F1值,综合评估批次矫正的效果。高的F1值来源于一个低的批次混合的ARI和一个高的细胞类型的ARI,最后通过Wilcoxon统计学检验和Benjamini Hochberg 矫正来检验这种方法是否是统计学最显著的批次矫正方法。
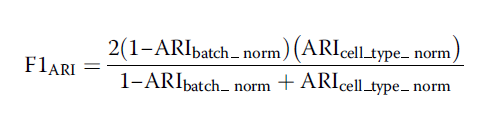
5.DEG (Differential gene expression analysis)
为了实现差异基因表达分析,作者在Seurat2中采用FindMarkers 函数通过似然比来检测的那细胞基因表达情况,采用Bonferroni correction(BF) 的方法来获取adjust 的p值,且阈值设置为0.05 。对原始的数据进行DEG 分析,它们包含批次效应,也对通过其他批次矫正后的矩阵进行DEG 分析。
DEG准确性分析
主要是TP,TN,FP,FN 和precision 的计算
接下来作者通过对不同的场景进行批次矫正处理
1.已知细胞类型,不同的技术
在这种情境下,在包含不同批次处理的有相同细胞类型的数据集中进行批次矫正检验。
那么首先 作者对数据集2小鼠细胞图谱进行批次分析和细胞类型分析,并通过UMAP进行可视化展示。结果如下
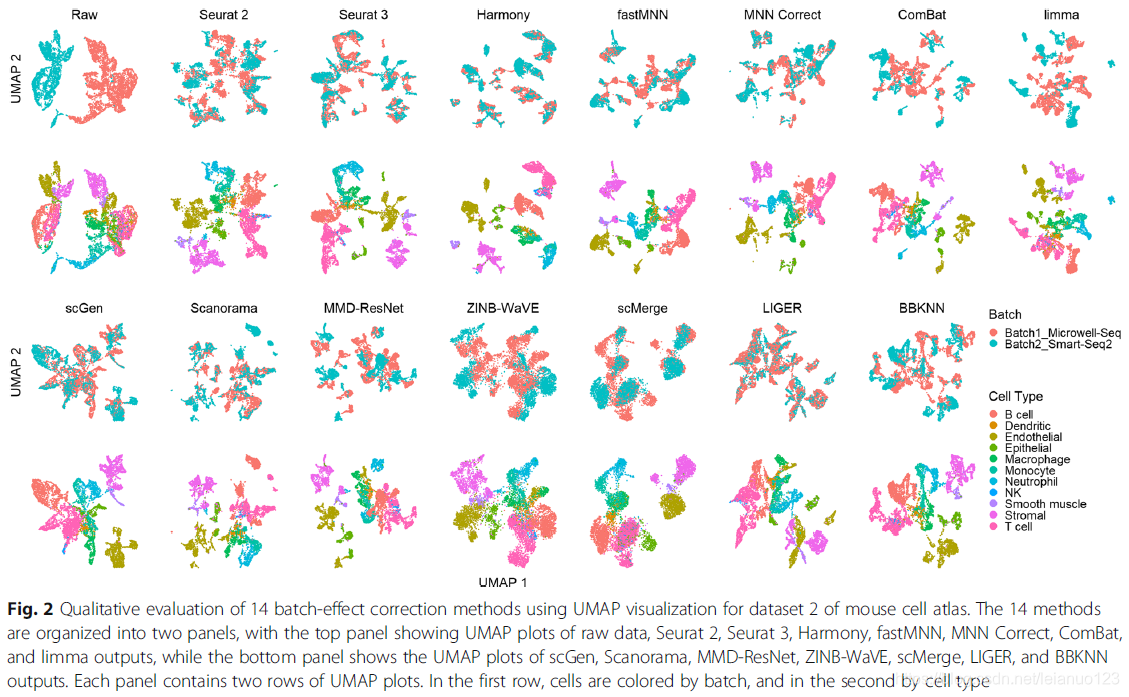 可以明显的看到,通过批次处理之后,数据集2中的数据能够充分混合重叠的是经过 Seurat 2, Seurat 3, Harmony, fastMNN, MNN Correct, scGen, Scanorama, scMerge,and LIGER successfully mixed the common cells这几个软件,同样的初看上去 这几个软件处理后的细胞类型聚类 也是这几个软件较好,能够相对较好的把每个细胞类型分开。ComBat ,limma,MMD-ResNet以及ZINB-WaVE以及BBKNN在相近的批次中能够产生相似的细胞类型。
可以明显的看到,通过批次处理之后,数据集2中的数据能够充分混合重叠的是经过 Seurat 2, Seurat 3, Harmony, fastMNN, MNN Correct, scGen, Scanorama, scMerge,and LIGER successfully mixed the common cells这几个软件,同样的初看上去 这几个软件处理后的细胞类型聚类 也是这几个软件较好,能够相对较好的把每个细胞类型分开。ComBat ,limma,MMD-ResNet以及ZINB-WaVE以及BBKNN在相近的批次中能够产生相似的细胞类型。
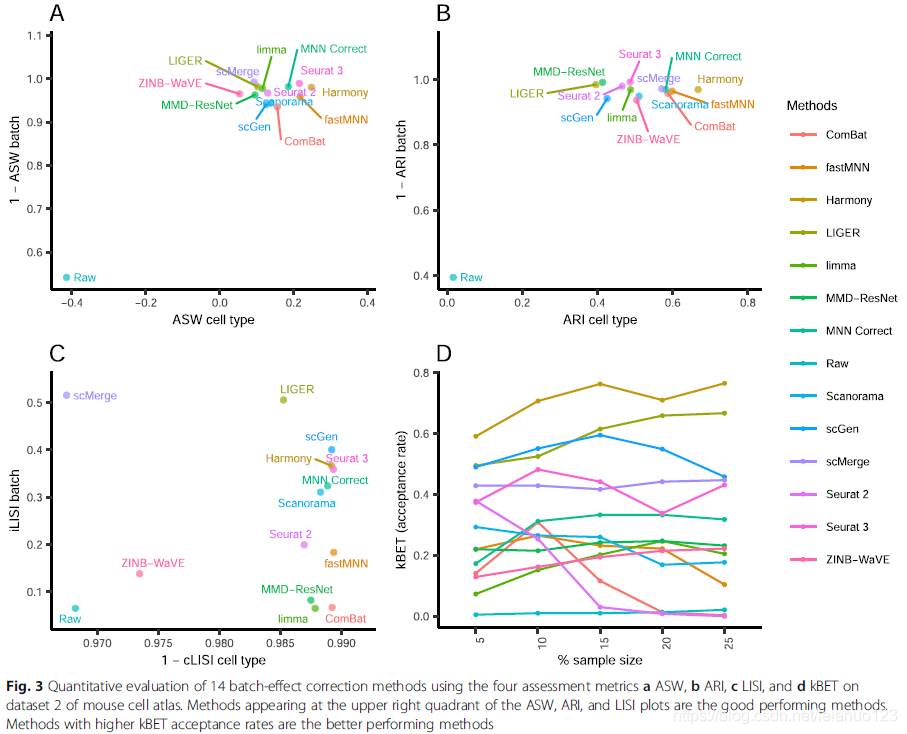
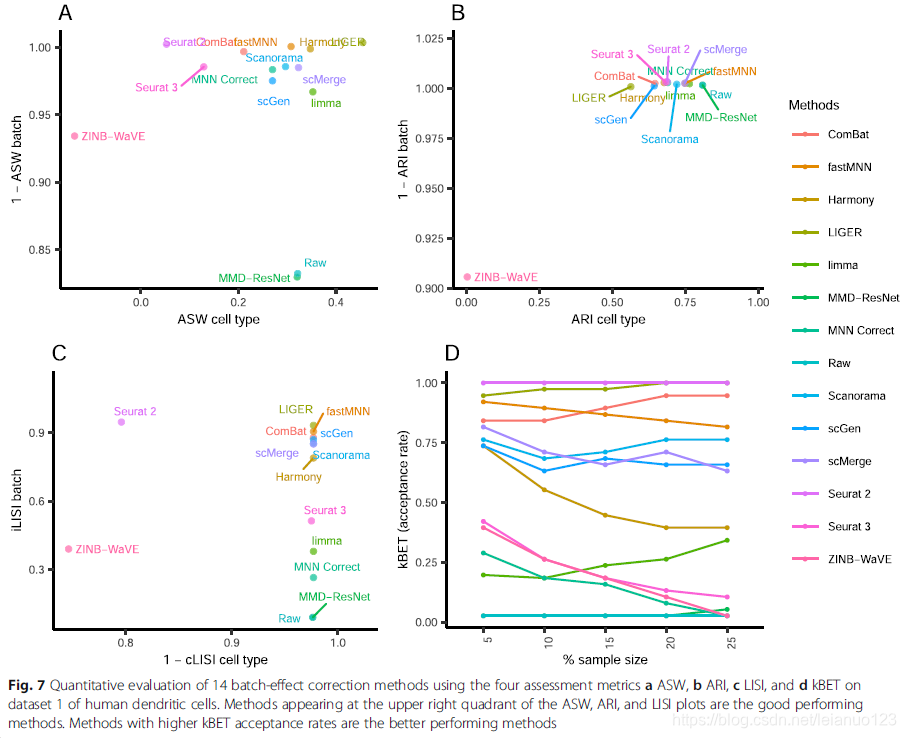
比较局部辛普森指数(iLISI)发现scMerge 是最好的方法用于批次混合,LIGER是第二好的方法,这两种方法的cLISI分数也非常好(1-cLISI)的值大于0.96,对于KBET,Harmony批次混合效果也非常好,接着是LIGER和scGen,p值都小于0.001。使用ASW来评估的话,Seurat3和Harmony 在批次混合和细胞聚类2方面都表现很好,其他的方法在批次混合效应上也很好,1-ASW的值>0.9,在ARI用于批次混合分析的时候,所有方法都获得了较好的ARI值,Harmony的p值为0.001,ARI的分数是0.67,在多种方法里,Harmony 排名第一,接着是MNN和Seurat3。
 对于第5个细胞集存在2对相似的细胞群,分别是CD4和CD8以及单核CD14和FCGR3A细胞,几乎所有的方法都无法分出这两个细胞簇,但是在Seurat2和3以及Harmony 和FastMNN 和MNN矫正过的数据里面,CD4和CD8亚群存在最小的细胞混合,在这些方法处理过后,这些亚群的分离是明显可见的。而scGen和MMD-ResNet以及LIGER也能混合批次,但是CD4和CD8细胞的混合也非常多。Scanorama ,ZINB-WaVE以及scMerge 不仅混合了CD4和CD8细胞,而且也批次也混合了。
对于第5个细胞集存在2对相似的细胞群,分别是CD4和CD8以及单核CD14和FCGR3A细胞,几乎所有的方法都无法分出这两个细胞簇,但是在Seurat2和3以及Harmony 和FastMNN 和MNN矫正过的数据里面,CD4和CD8亚群存在最小的细胞混合,在这些方法处理过后,这些亚群的分离是明显可见的。而scGen和MMD-ResNet以及LIGER也能混合批次,但是CD4和CD8细胞的混合也非常多。Scanorama ,ZINB-WaVE以及scMerge 不仅混合了CD4和CD8细胞,而且也批次也混合了。
cLISI特点,发现细胞类型纯度高达0.98,但是cLISI这一特点只用于分析局部的细胞纯度,特异细胞类型边缘的混合效果不好。LIGER在批次混合上位于第一,而Seurat2和3位于其后,这与KBET计算的结果一致。除了ASW特点外,所有的批次混合值都大于0.95,而Harmony 和Seurat3都排名于top,且p值为0.183,接着是MNN。和ARI一样,就细胞类型的纯度而言,Harmony 排名第一,接着是fastMNN和Seurat3和MNN,折4种方式的ARI的批次分数都大于0.97,使用rank sum .Harmony和Seurat3和LIGER排名前三。
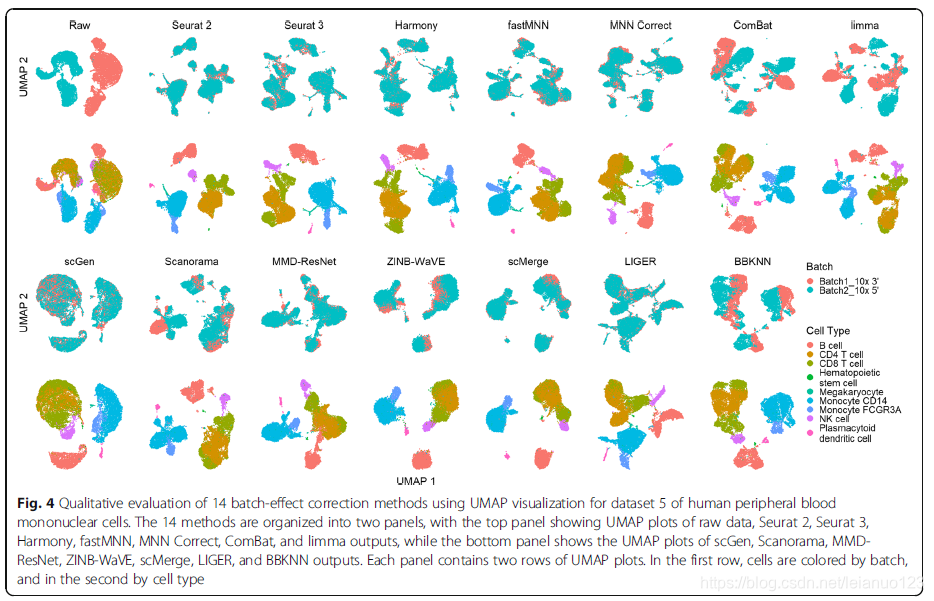
对于所有的数据集来说,Harmony 都是排名第一,其次作者推荐的几个方法是Seurat3和LIGER和FastMNN。
场景二
不确定的细胞类型
在数据一中,出现的都是高度相似的2个细胞类群,通过不同的批次矫正的方法发现几乎所有的方法都能够将2个批次的细胞很好的混合在一起。limma 包能够将2个细胞cluster 聚集在一起,但是无法很好的将2个批次混合起来,然而MMD-ResNet和BBKNN却不能很好的将细胞cluster 聚集在一起。 多数批次矫正算法,都要求至少有一个细胞群在2个数据集间共享,从而去指导数据的比对,其中MNN和FastMNN和Seurat3和Scanorama都通过寻找MNN来寻找2个数据共有的细胞类群,而当2个数据间没有共有的细胞亚群的时候MNN的错误匹配以及不正确的比对将会发生,尤其是2个数据集中的细胞亚群高度相似的时候,这也是为什么批次矫正的方法都将CD1C和CD141细胞整合到了一起。
多数批次矫正算法,都要求至少有一个细胞群在2个数据集间共享,从而去指导数据的比对,其中MNN和FastMNN和Seurat3和Scanorama都通过寻找MNN来寻找2个数据共有的细胞类群,而当2个数据间没有共有的细胞亚群的时候MNN的错误匹配以及不正确的比对将会发生,尤其是2个数据集中的细胞亚群高度相似的时候,这也是为什么批次矫正的方法都将CD1C和CD141细胞整合到了一起。
就KBET分数来说,LIGER 和Seurat2对数据集的批次整合效果都很好,LIGER和seurat2 都能够很好的进行批次整合,就iLISI 检测结果来说,LIGER和Seurat 2都能够得到很高的分值。
数据6只包含2种细胞类型,而2/3的细胞都只含有一种细胞类型,而t-SNE和UMAP表明scGen,scMerge 以及BBKNN都能够产生2个大型的特异的细胞类群293T和Jurkat,Harmoy 也能够很好的进行批次混合,能够将两类细胞分为2组,LIGER也能够产生批次混合的细胞簇,但是在一些细胞类型上存在混合,但是在Seurat2的输出结果里,2种细胞类型的点靠得很近,难以分开。从LISI特点表明Harmony,scMerge 以及scGene都是最好的方法,从批次混合何细胞类型纯度来说
293T和Jurkat细胞的混合和聚类情况
 场景3多批次
场景3多批次

场景4大数据
BBKNN,ComBat, Harmony, LIGER, limma, MMD-ResNet, Scanorama,
scGen, Seurat 3, and ZINB-WaVE这几个算法都能够跑全库,而其他的几种算法不能,其中FastMNN 和scMerge以及Seurat2等算法,需要的内存高达2。27TB,而MNN 算法进行批次矫正的时长约要48h以上。
以上的那些算法中,LIGER能够很好的维持细胞类型的分离,从而实现批次混合,Seurat 3,Harmony, ZINB-WaVE, scGen, and MMD-ResNet效果相对较差,而Limma 和Combat 以及Scanorama,BBKNN这几种算法实现批次混合的效果很差,几乎没有批次的混合。总之,scGen是最好的方法,批次混合的p值小于0.001,并于LIGER一起细胞类型纯度达到0.34。
场景5数据的模拟
批次矫正分析,主要是为了后续下游的伪时序以及基因差异分析。在此方法张作者通过综合分析8种方法批次矫正后的基因差异分析。作者选用具有批次
效应样本的全部基因或者高变异基因作为批次矫正的输入数据,批次校正后,进行DEG分析,最后通过TP,TN以及FP,FN来计算DEG 的准确率。那么作者通过计算发现,在使用HVG作为批次矫正的关键基因时,进行批次矫正后能够得到更真实的DEG,具体的见图20C。
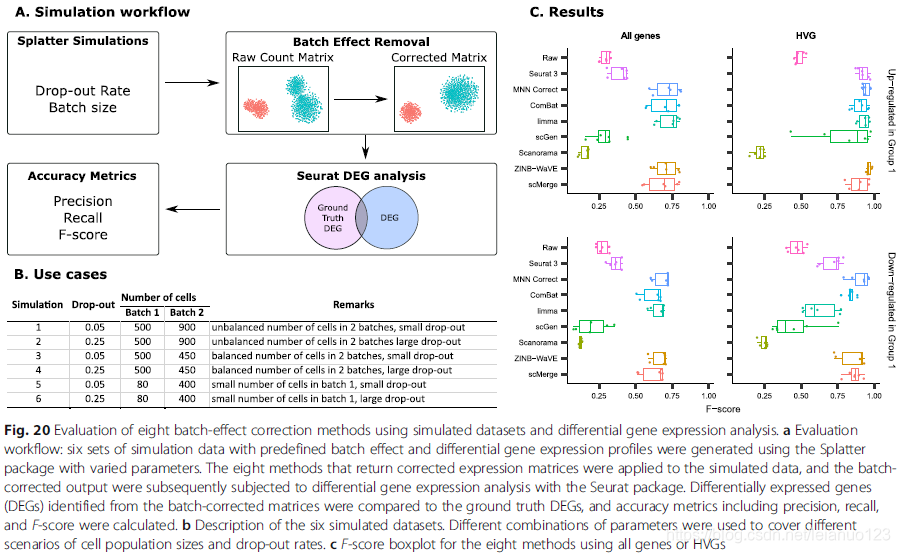 那么如何解释HVG对于批次矫正后,能够更好的进行后续差异分析,主要是因为HVG能够更好的保留细胞类型自身的特点,因此使用选择的HVG进行批次矫正能够提供更好的精确性,但是使用HVG 限制了后续的分析,因为基因数目有限。因此有必要通过全部的基因进行批次矫正处理,然后在得出结论的时候仔细小心以防出现假阳性或者假阴性FP或者FN。
那么如何解释HVG对于批次矫正后,能够更好的进行后续差异分析,主要是因为HVG能够更好的保留细胞类型自身的特点,因此使用选择的HVG进行批次矫正能够提供更好的精确性,但是使用HVG 限制了后续的分析,因为基因数目有限。因此有必要通过全部的基因进行批次矫正处理,然后在得出结论的时候仔细小心以防出现假阳性或者假阴性FP或者FN。
通过场景5作者推荐用于后续的DEG分析的方法主要包括ComBat,MNN以及ZINB-WaVE以及scMerge。
讨论
1.批次矫正的方法
2.批次整合评估方法包括KBET iLISI,ARI和ASW。对于前2者来说,两者的一致性非常高主要是二者都是基于邻域的,而对于ARI的准确性来说,主要依赖于聚类算法和簇的数目
3.运行时间和内存需求 推荐最佳的方法还是Harmony,LIGER以及Seurat3,对于大数据集的处理,Harmony 处理时间低于1h
结论
通过14中批次矫正的方法和5种情景,作者发现综合考虑,在进行单细胞的批次处理的时候,LIGER,Harmony 以及Seurat3都能够很好的进行批次处理。Harmony 无论是对常见的细胞类型以及不同的技术和运行时间上都是非常不错的方法,同样LIGER也是,尤其在未知细胞类型的时候,对于LIGER来说主要的缺陷是运行时间长,Seurat3也能处理大数据集,但是其运行时间笔LIGER还要长20%-50%,然而对于若是进行下游的DEG分析,作者推荐scMerge进行批次矫正。
全文用到的批次矫正方法

















 1万+
1万+

 被折叠的 条评论
为什么被折叠?
被折叠的 条评论
为什么被折叠?
 到【灌水乐园】发言
到【灌水乐园】发言









