






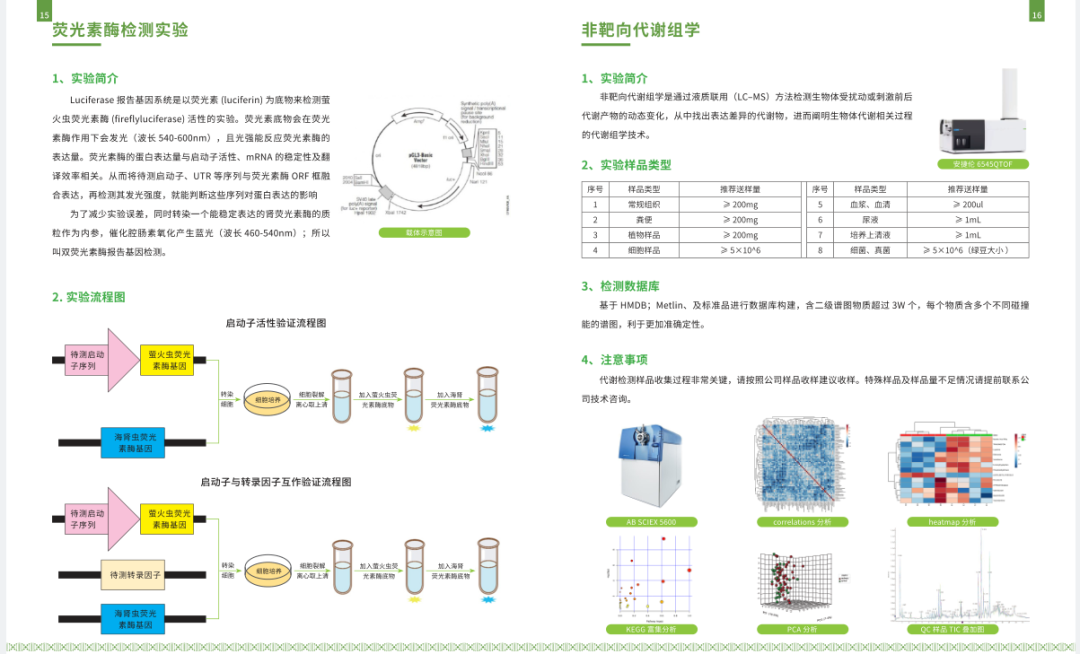
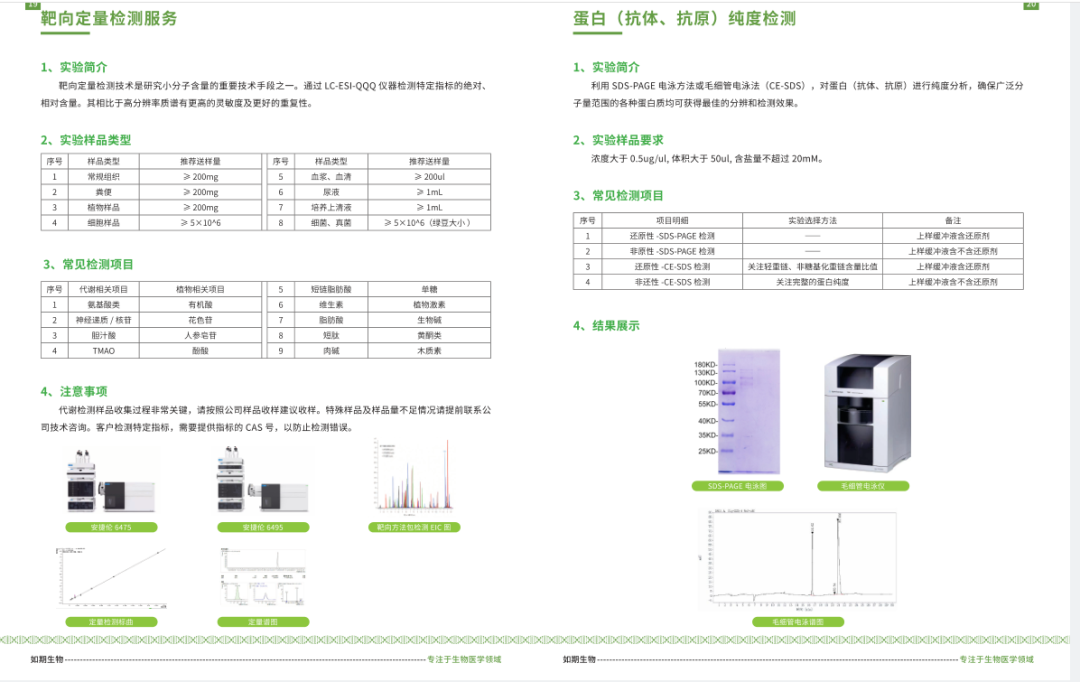
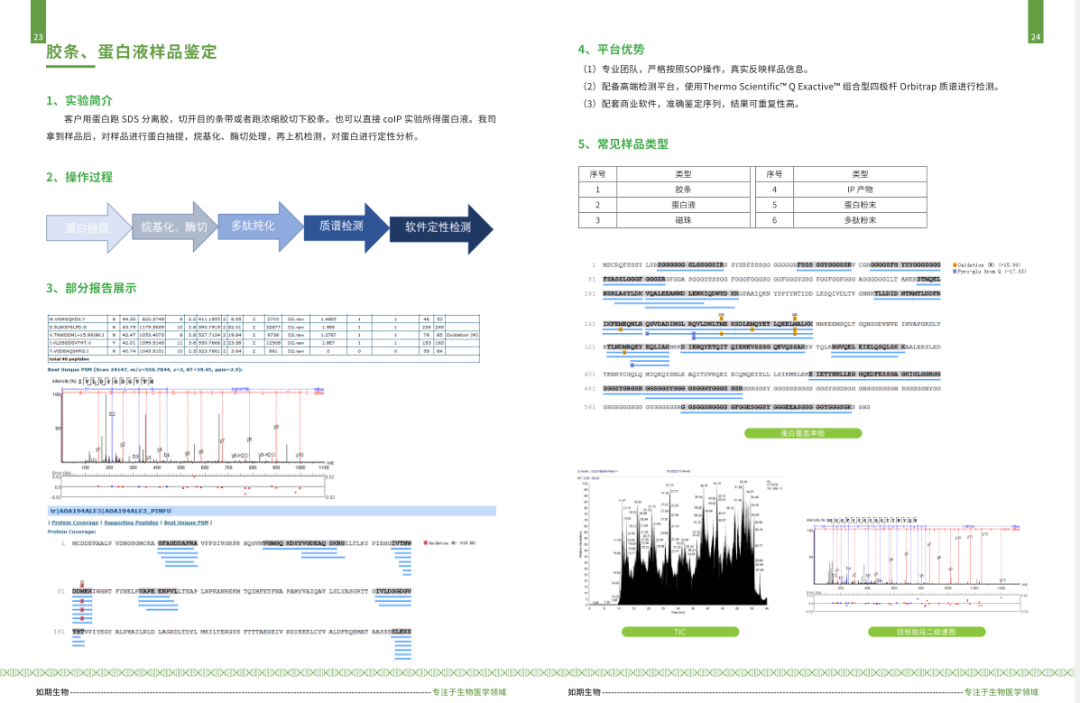
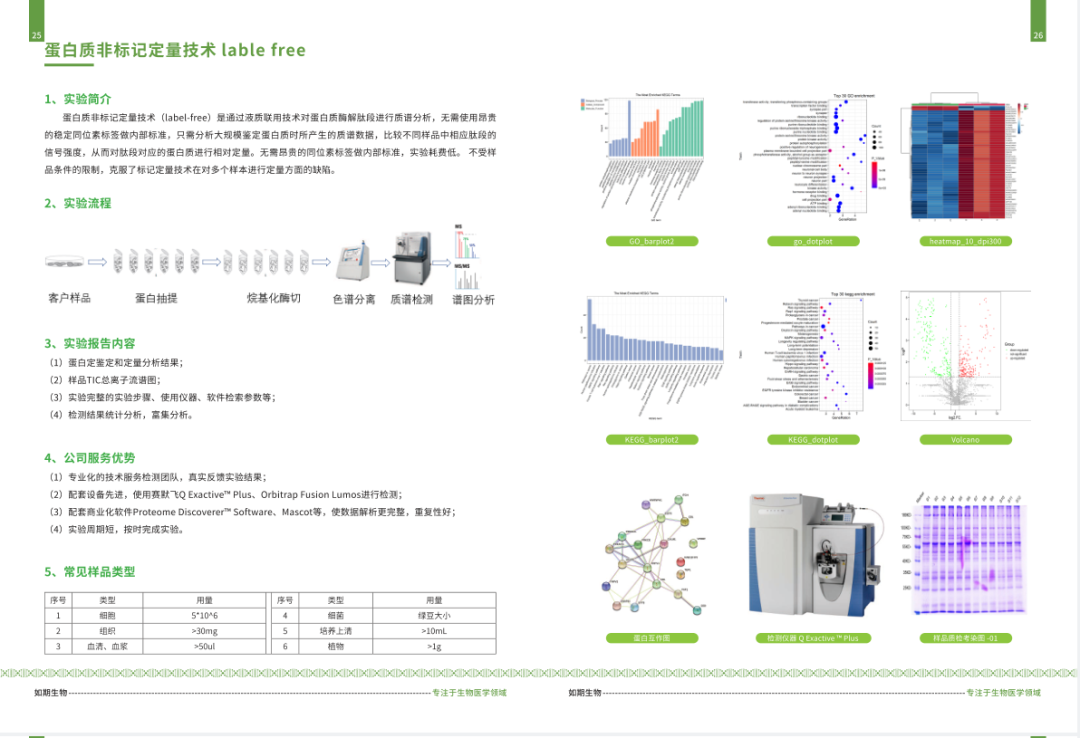
启动子(promoter)是与RNA聚合酶及转录因子结合调节基因表达的序列。
下边我们以人的“FAU”为例介绍一下启动子的查找:
网址:http://genome.ucsc.edu/index.html
1.打开网页,如下图选择左上角下拉菜单的“Human GRCh38/hg38”
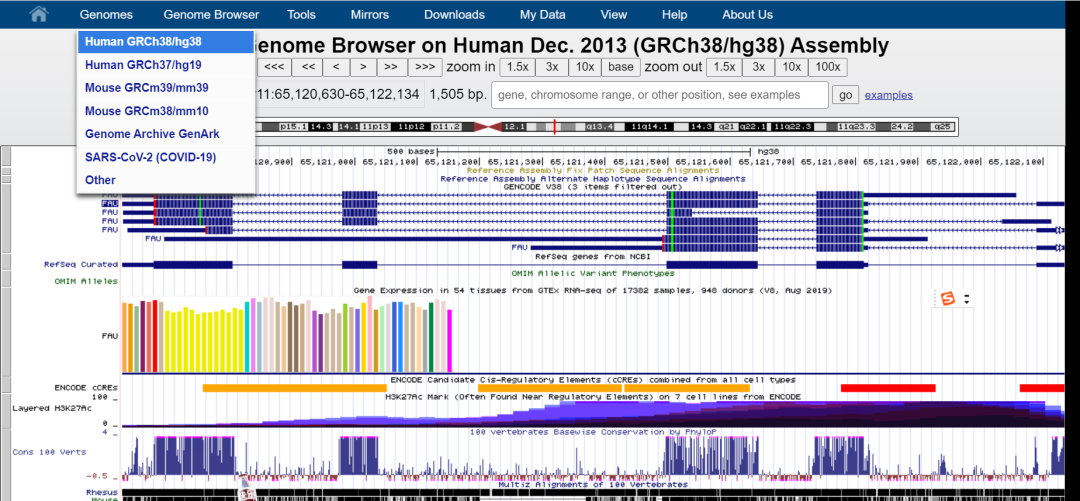
2.输入需查找的基因"FAU",点击go:
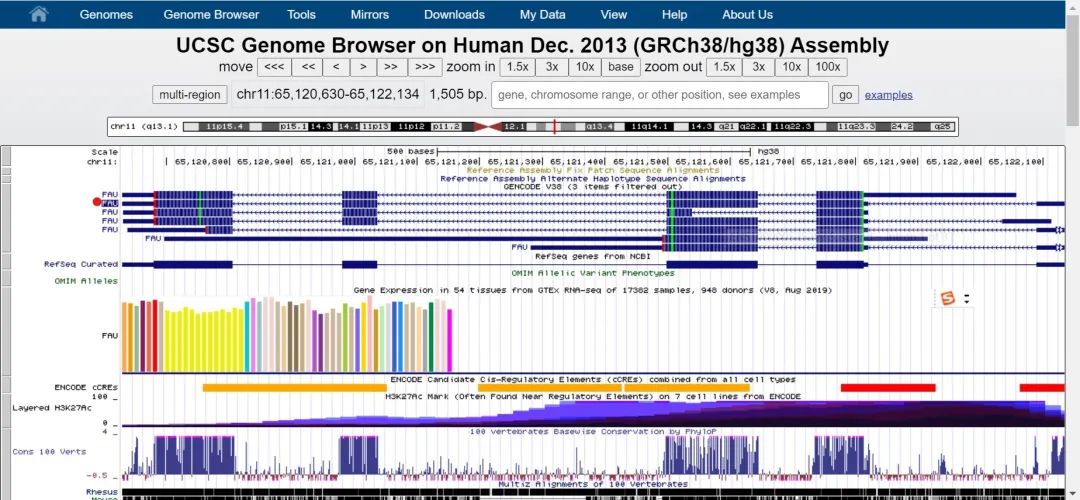
2.选择目的序列,并单击;跳转到如下界面;
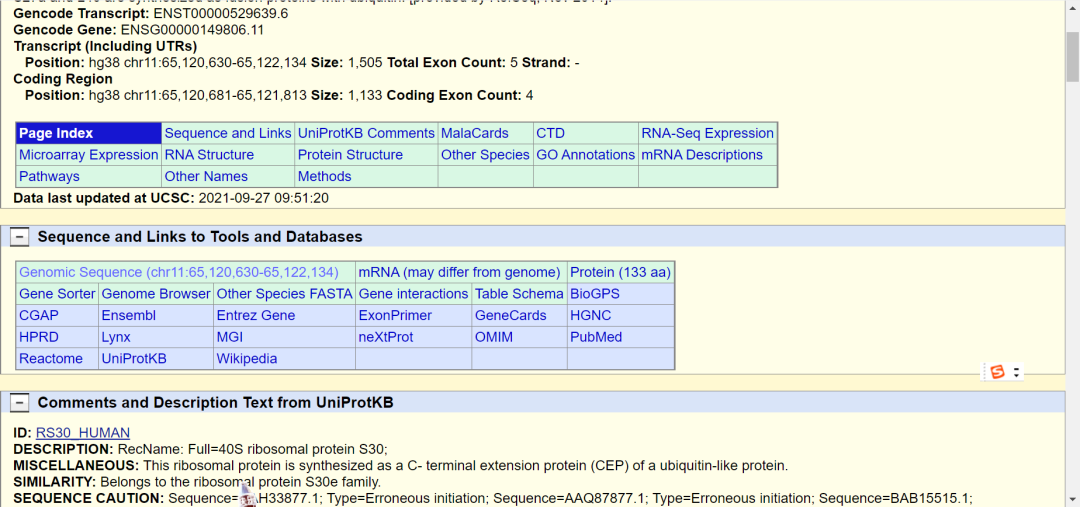
3.点击红色框:
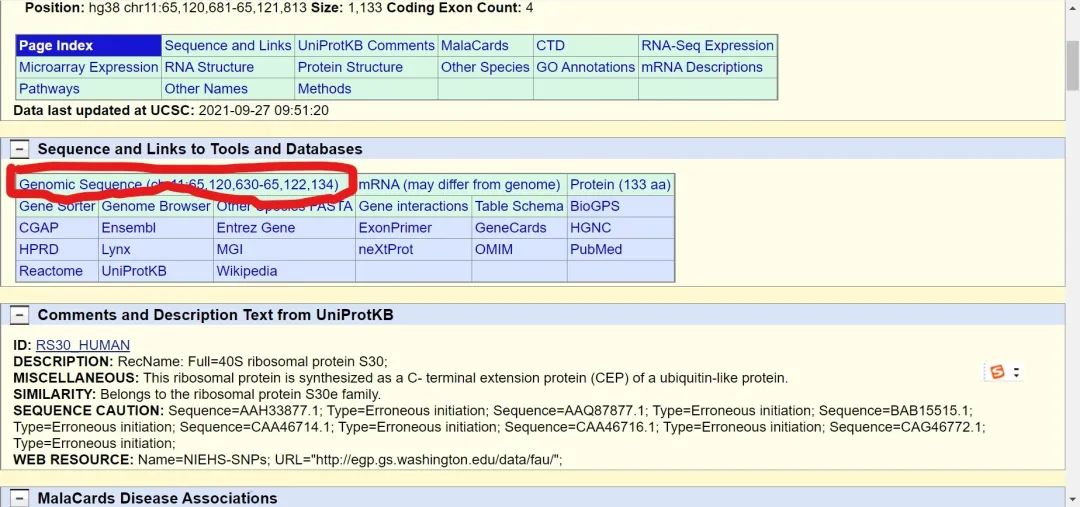
4.按下图所示选择输出参数;并点击“submit”:
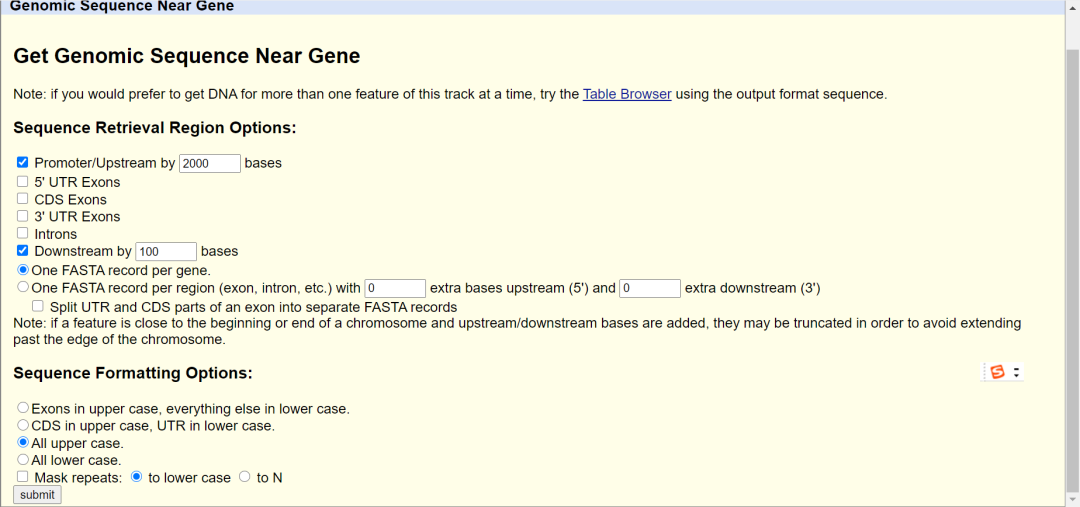
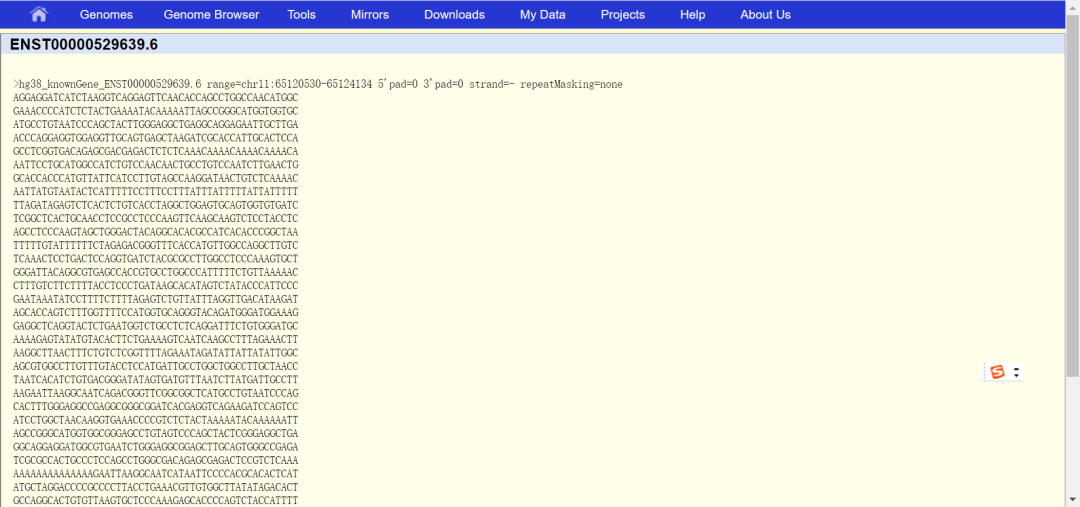
5.最终查询到的启动子信息如下图所示:
如各位研友,在实操过程中遇到什么问题可以通过下方公众号的联系方式与我们一起探讨交流。
PS: ChIP-qPCR 可用于研究已知基因组结合位点处的蛋白质-DNA相互作用。如果位点未知,qPCR引物也可以针对潜在的调控区域(例如启动子)进行设计。ChIP-qPCR在关注特定基因和潜在调控区域的研究中具有优势,因为qPCR的成本极低,可以在不同的实验条件下进行。ChIP-qPCR技术实现了在靶基因的启动子上找到转录因子结合的直接证据,是细胞内真实的、原位的结果,同时可以比较与不同位点的结合能力的比较,相对于其他体外实验验证更具说服力。


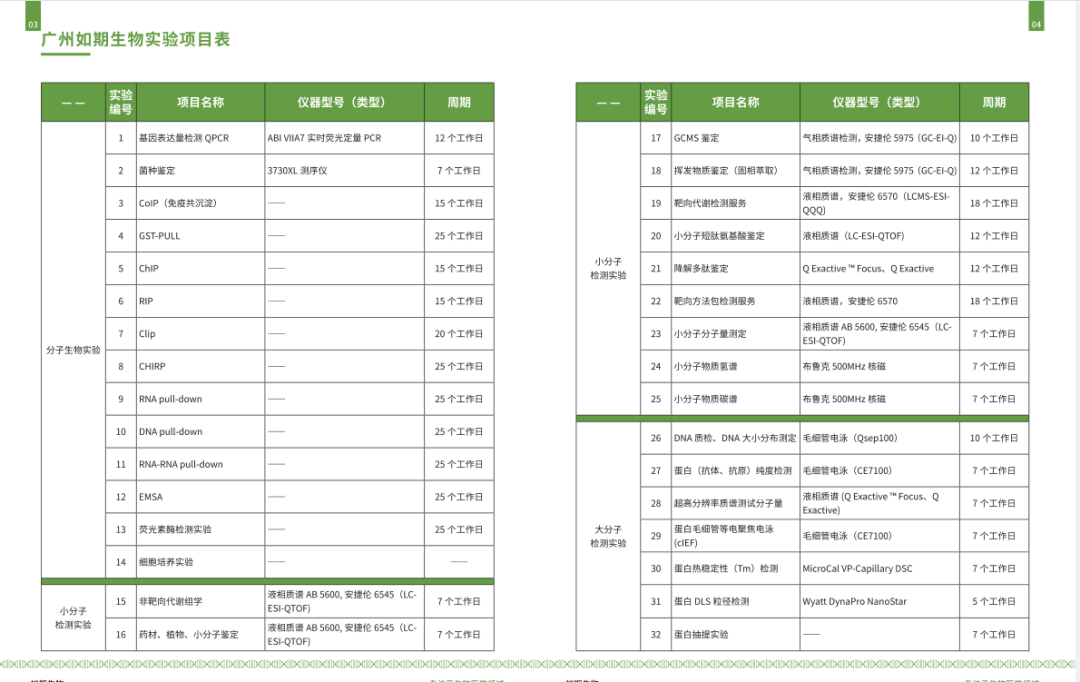


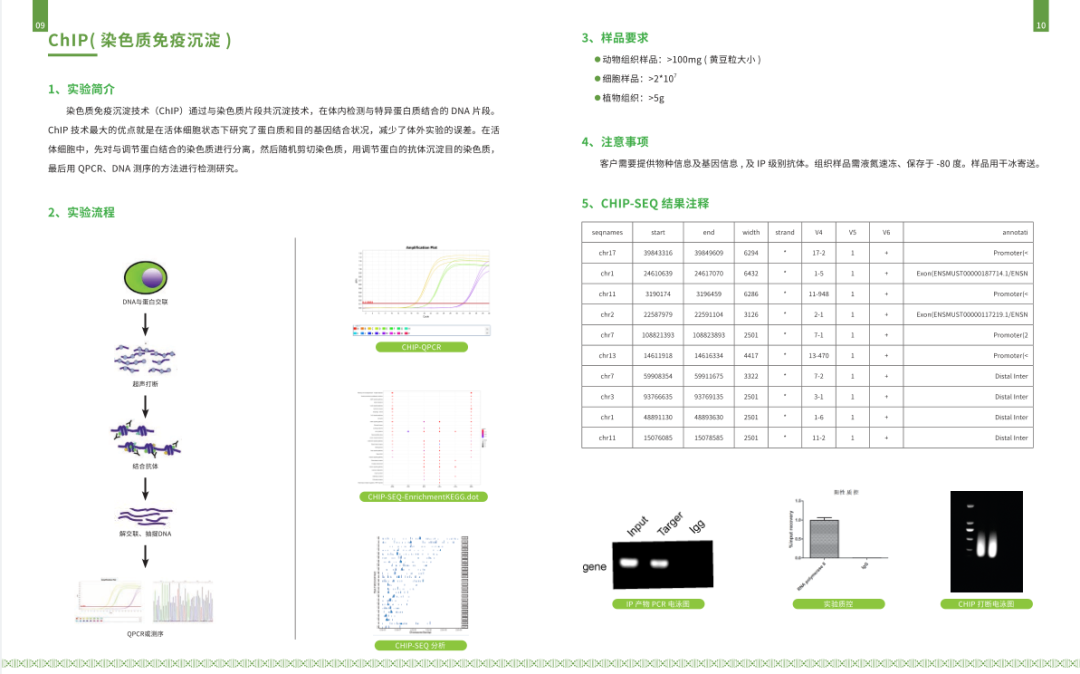























 1万+
1万+

 被折叠的 条评论
为什么被折叠?
被折叠的 条评论
为什么被折叠?
 到【灌水乐园】发言
到【灌水乐园】发言











