







1. 导入蛋白质
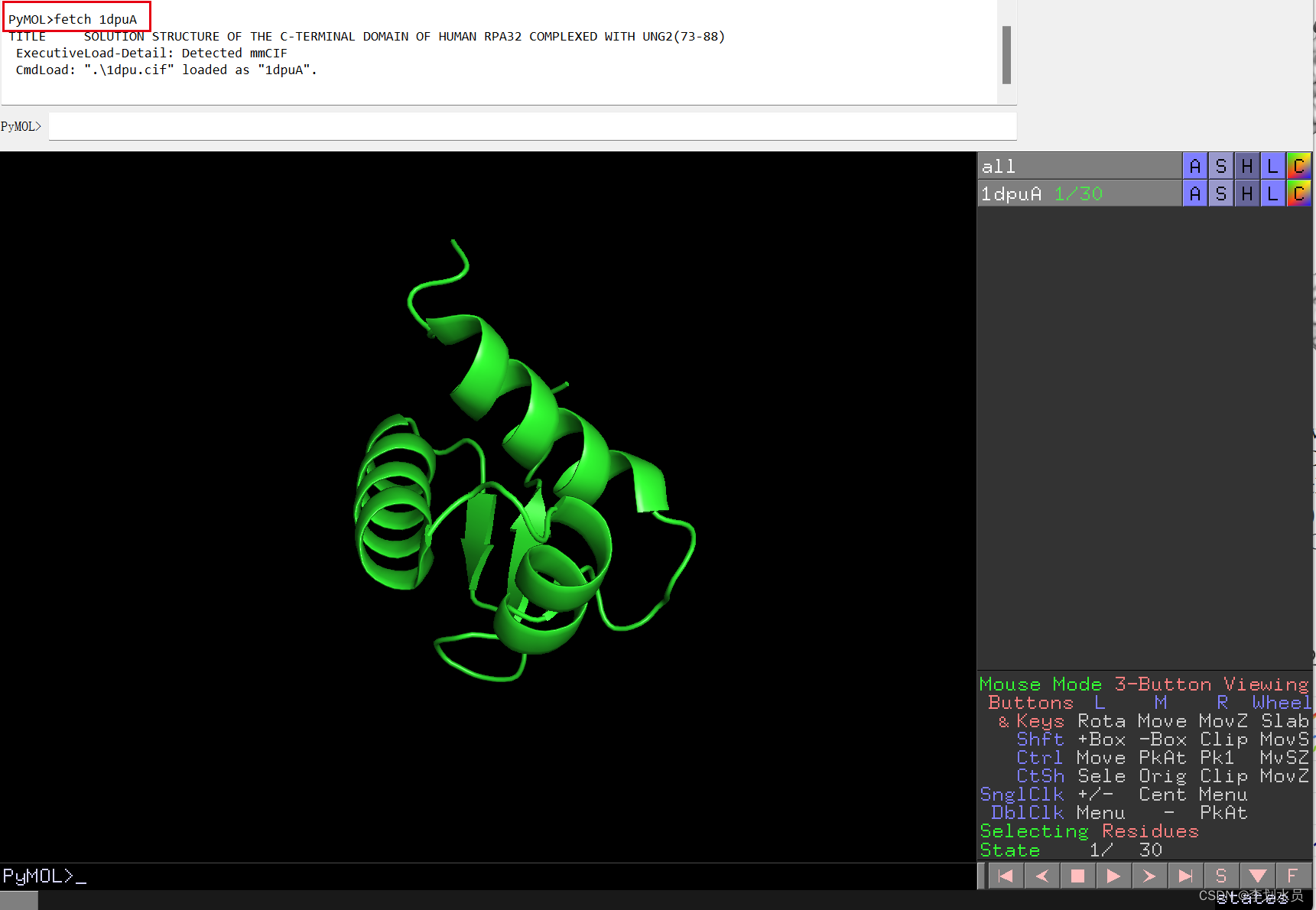
1)Pymol> load name.pdb, name # 载入pdb文件,并命名,我还没试过
Pymol> fetch proteinID # 直接就加载了 我用的这个
右边选框,有A S H L C指令
2. 保存图片
- 2.1 直接输出PNG,在pymol后输入:
![]()
png name.png进入Script文件查看保存测文件,路径在上一篇的pymol

2.2 设置PNG的参数(仅记录,未实践)
png filename [, width[, height[, dpi[, ray[, quiet]]]]]- filename = string(字符串): 文件名称和文件的路径。
- width = integer or string (整数或字符串): 宽度,单位为像素。如果给定单位后缀英尺或厘米(in, cm),则需要dpi参数。如果只给出 width或 height中的一个,则保留可视化窗口的长宽比,默认是0。
- height = integer or string(整数或字符串): 高度,默认是0。
- dpi = float: dots-per-inch 默认为 -1.0 。
- ray = 0 或 1: 如果为1则在输出png前运行ray,默认是0。
- quiet = 0 或 1: 如果为1则不输出log日志,默认是 0。
学习文章:















 2855
2855











 被折叠的 条评论
为什么被折叠?
被折叠的 条评论
为什么被折叠?
 到【灌水乐园】发言
到【灌水乐园】发言









