









1、需要软件:autodocktools,vina,pymol
pymol使用教程(包含安装):基础教程 — PyMOL中文教程 2022.09 文档
autodocktools及vina安装包:
「autodocktools」https://www.aliyundrive.com/s/88pQFmFBNKK 点击链接保存,或者复制本段内容,打开「阿里云盘」APP ,无需下载极速在线查看,视频原画倍速播放。
2、需要蛋白质文件及小分子文件
需要文件为蛋白质文件:.pdb(PDB上下载,或直接在pymol上处理:fetch ####)
小分子配体文件:.mol2格式或.pdbqt格式(利用openbabel进行格式转换)
简单介绍一下openbabel:
openbabel是化学信息学中常用的一款格式转换的工具,用openbabel可实现smi,mol2,pdbqt等多种格式之间的相互转换。
Open Babel由C/C++编写,并提供C ++, Perl, Python等的API接口方便开发。
想深入了解openbabel的,请参考下面的文献:
 https://jcheminf.biomedcentral.com/articles/10.1186/1758-2946-3-33
https://jcheminf.biomedcentral.com/articles/10.1186/1758-2946-3-33下面演示openbabel的安装及简单的使用demo:
# anaconda中openbabel的安装(windows)
conda install openbabel -c conda-forge
# 验证安装成功——显示版本号
obabel -V
'''openbabel可用于文件格式转换,过滤(按smarts,标题,描述符值或属性字段), 3D
或2D结构生成,氢从隐式转换显式,并删除小片段或重复结构'''
# 最常见使用场景将smiles格式转化为sdf格式
obabel -ismi imatinaib.smi -osdf imatinaib.sdf
# 在转换过程中去掉所有氢
obabel -isdf imatinaib.sdf -osmi iamtinaib.smi -d
# 在转换过程中加上所有氢 ———— 一般不建议在openbabel过程中加氢
obabel -isdf imatinaib.smi -osmi imatinaib.smi -h
# 在转换时添加合适pH=7.4的氢
obabel -isdf imatinaib.sdf -osmi imatinaib.smi -p
# 添加所有氢但不更改文件类型
obabel -isdf imatinaib.sdf -osdf imatinaib.sdf -h
# 一些官能团,例如硝基或砜,可以表示为[N+]([O-])=O或N(=O)=O,以将全部转化为配位键形式
obabel -isdf imatinaib.sdf -osmi imatinaib.smi -b
# 转换分子的子集,可以使用-f和-I定义它们,demo:要转换文件mymols.sdf类型的分子2-4
obabel mymols.sdf -f 2 -l 4 -osdf outputfile.sdf
# 筛选与SMARTS模式匹配的子集,demo:选择出所有含溴苯的分子
obabel mymols.sdf -osdf 'selected.sdf' -s 'c1ccccc1Br'
# 筛选与SMARTS模式不匹配的子集,demo: 选择出不含溴苯的分子
obabel mymols.sdf -osdf 'selected.sdf' -v 'c1ccccc1Br'产生三维结构的时候顺带通过MMFF94高精度有机小分子力场进行优化,demo: 运行如下命令,得到的pdbqt文件里的结构就都是优化过的了
obabel -ismi imatinaib.smi -opdb imatinaib.pdb --gen3d -m --minimize -ff MMFF943、处理大分子蛋白及有机小分子demo
大分子蛋白处理过程:
在处理蛋白前,先做准备工作,在PDB及PDBSum中搜索蛋白结构,下面以有蛋白配体复合物晶体结构的一个样本为例:1SM2与星型孢菌素(STU)结合物
在PDBSum中可以得到一些关于1SM2的信息:
prot-prot中可得到chain A 与chain B是等效的,两条链通过氢键连接到一起,我们在对接时选择其中的一条处理即可。
Ligand中可以得到蛋白与STU作用的方式及位点,可以看到主要相互作用为氢键和疏水作用,STU与Arg(486),Glu(436),Met(438)之间分别有较强的氢键相互作用(键长3埃米以下)。
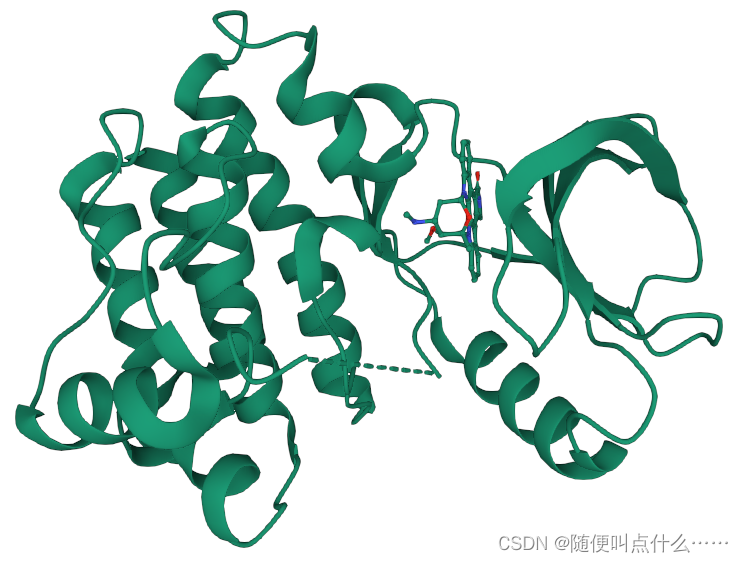
首先,处理蛋白质分子,打开pymol
# 打开要处理的蛋白结构
fetch 1SM2 ####(蛋白的四位编码,如:1SM2)
# 除去所有非键合水
remove resn HOH
# 删除所有与对接无关的配体分子,金属离子等
# 1SM2两条链是等效的,我们处理其中一条
create 1SM2A, Chain A
# 方便操作,删掉不需要的对象
delete 1SM2
# 将复合物的配体复制为一份单独的对象
create ligand, RESN STU
# 将复合物的蛋白质复制为一份单独的对象
create protein, 1SM2A and not RESN STU
# 保存蛋白为本地PDB文件
save d:/untitled/protein.pdb protein
# 保存配体为本地PDB文件
save d:/untitled/ligand.pdb ligand
根据对蛋白配体结合物的分析,氢键长度范围在2.5-3.0埃米之间,将配体STU周围5埃米残基定义为结合位点并创建对象
# active是选择的命名,sele要选的对象
select active, byres all within 5 of ligand这一步可以对结合位点进行更好的渲染,非必须。
在autodocktools中打开protein.pdb以及ligand.pdb两个文件,进行加氢—合并非极性氢—电荷优化等操作(在电荷优化中,autodock工具中默认的一些金属离子,如Mg,Zn等默认带电荷为零,这里需要手动在.pdbqt文件中进行更改电荷参数)。
加氢:Edit—Hydrogens—Add—OK
合并非极性氢:Edit—Hydrogens—Merge Non—Polar
进行电荷优化:Edit—Charges—Computer Gasteiger (Gasteiger是电荷优化计算的一种,轨道电负性部分均衡(PEOE)电荷,这种方法运用了电负性均衡概念,即电负性不同的原子成键时,电负性较小的原子附近电子密度会流向电负性较大的原子。在这个过程中原先电负性小的原子电负性会增大,当所有原子间电负性相等时,电子密度非分布就是平衡状态分布。实际上PEOE电荷并没有达到完全的电负性均衡,而只达到了部分均衡)
处理蛋白后将其保存成pdbqt格式,方面后面使用。
小分子的处理过程:
redocking过程:什么是redocking,如果你的受体蛋白在PDB数据库里有和其它小分子复合的结构,你可以先做这个小分子对接。然后,把对接的结果和原来的真实结构重叠比较看一下这个对接软件在你的受体蛋白上表现怎么样,是不是可以准确预测结合方式。
对于redocking的过程,只需要处理单个小分子,在ZINC或TCMSP官网下载小分子mol2格式,用pymol打开保存为.pdb格式(或者也可以用从PDB蛋白小分子配合物中提取出来的.pdb文件,都是可以的)。
预处理过程:使用autodocktools预处理小分子。首先,读入小分子:file—read molecule(导入小分子pdb文件);加氢:Edit—hydrogens—Add;计算电荷:Edit—charge—compute gasteiger;指定为对接小分子:ligand—input—choose—选中小分子—select molecule for autodock4;Ligand—torsion tree—detect root-torsion tree—choose torsions-done;输出:ligand—output—save as pdbqt。
Vina对接过程:
打开处理好的大分子pdbqt结构:Grid—Macromolecule—open(打开大分子.pdbqt文件)。变成二级结构看起来会比较美观:display—Secondary Structure—display二级结构的形式展示。
打开处理好的小分子配体pdbqt结构:Grid—Set Map Types—open ligand(打开你的小分子.pdbqt文件)。
建立对接盒子:Grid—Grid Box,选择盒子中心:Center—Center on ligand—File close saving current,保存对接盒子的参数:Grid—output—Save GPF
写config文件:建立一个config.txt文件,内容参考如下:
receptor = protein.pdbqt
ligand = molecule.pdbqt
# 注释:center部分的内容填写参考GPF
center_x = 24.273
center_y = -4.659
center_z = 28.987
# 注释:size部分的内容填写也参考GPF
size_x = 40
size_y = 40
size_z = 40
energy_range = 4
out = molecule_out.pdbqt
log = molecule_out.pdbqt
# 注释:exhaustiveness是一个精度相关的参数,数值越大,时间开销越大,精准度也越高
exhaustiveness = 24
命令行操作:
vina --config config.txt --out result.pdbqt运行后等待查看结果即可。
对于redocking的过程,主要看对接的均方根偏差RMSD(Root Mean Square Deviation)值与单晶结构中的配体接近程度,越接近,对接能量越低则越好。当对接构象与晶体构象间的重原子RMSD值<=2埃米时,认为对接成功,反之则为失败(所有RMSD值可采用Schrodinger程序重的rmsd.py脚本计算)。
下一篇将介绍批量小分子对单一蛋白靶点的对接。

















 2291
2291

 被折叠的 条评论
为什么被折叠?
被折叠的 条评论
为什么被折叠?
 到【灌水乐园】发言
到【灌水乐园】发言









