









用R包Hmisc里的rcorr函数计算相关性,并将输出矩阵格式进行转换。
 这里我用的是单表,行为样本名,列为细菌相对丰度,想计算细菌之间的相关性。rcorr函数计算相关性的方法有两种:Pearson和Spearman,使用示例如下:
这里我用的是单表,行为样本名,列为细菌相对丰度,想计算细菌之间的相关性。rcorr函数计算相关性的方法有两种:Pearson和Spearman,使用示例如下:
Usage
rcorr(x, y, type=c(“pearson”,“spearman”))
install.packages('Hmisc')
library(Hmisc)
data<-read.table("clipboard",header = T,row.names = 1)
res<-rcorr(as.matrix(data))
flattenCorrMatrix <- function(cormat, pmat) {
ut <- upper.tri(cormat)
data.frame(
row = rownames(cormat)[row(cormat)[ut]],
column = rownames(cormat)[col(cormat)[ut]],
cor =(cormat)[ut],
p = pmat[ut]
)
}
final<-flattenCorrMatrix(res$r,res$P)
write.table(final, 'stage', sep = '\t', col.names = NA, quote = FALSE)
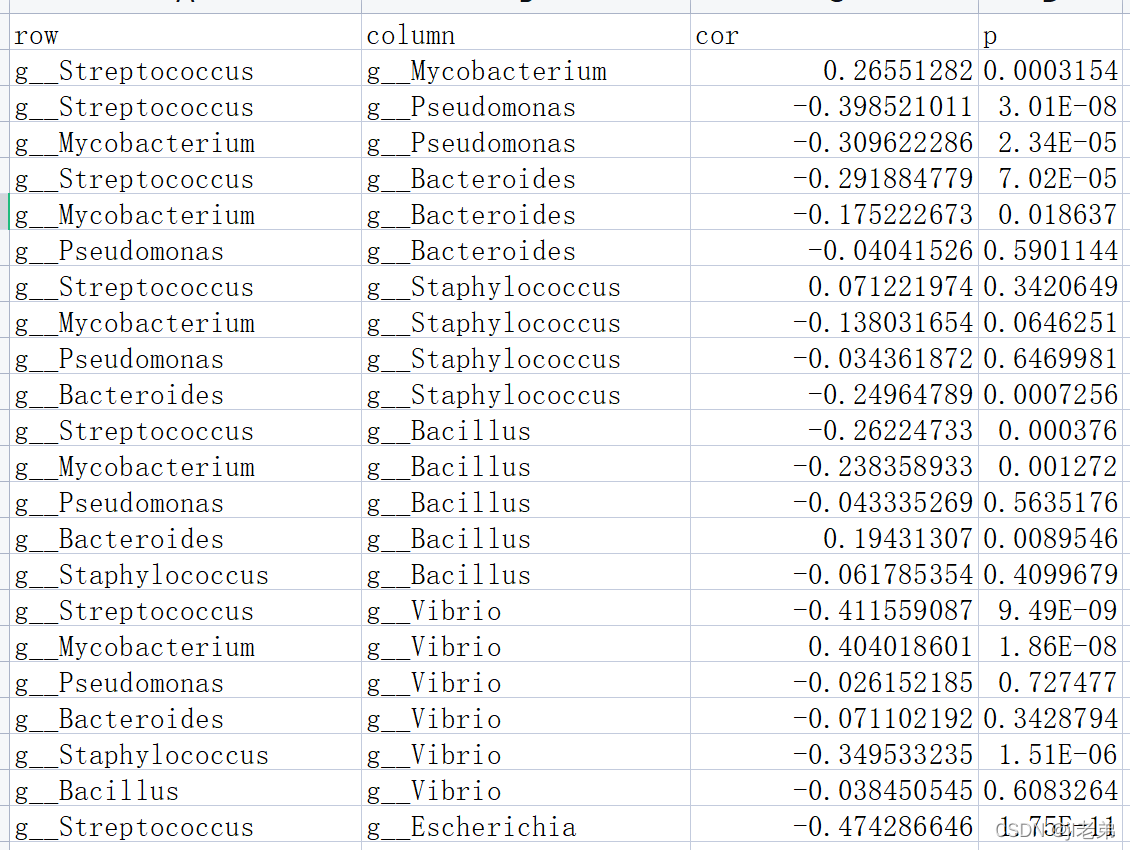
我的结果是细菌之间的相关性,输出第三列是相关性系数,第四列是p-value;对相关性系数和p-value进行筛选后,可以导入Gephi或者cytoscape进行网络图的绘制。

















 371
371

 被折叠的 条评论
为什么被折叠?
被折叠的 条评论
为什么被折叠?
 到【灌水乐园】发言
到【灌水乐园】发言









