






- 动力学模拟轨迹转为pdb格式命令,从而用pymol分析
gmx trjconv -f md.xtc -s md.tpr -n index.ndx -o md_cluster_whole.xtc -pbc cluster -center
gmx trjconv -f md_cluster_whole.xtc -s md.tpr -n index.ndx -fit rot+trans -o md_cluster_fit.xtc
gmx trjconv -f md_cluster_fit.xtc -o md_cluster_skip.xtc -skip 100
gmx trjconv -f md_cluster_skip.xtc -s md.tpr -o md_cluster_skip.xtc
- pymol程序安装启动后,点击File > Open > md_cluster_skip.xtc,就能得到下图,红色框内State表明这个pdb文件中有多少帧构象,点击红色框内最下面一行的按钮就可播放动力学模拟轨迹。
在PyMol Viewer中出现all、md两栏,每栏都有A、S、H、L、C,A:Action,S:show,H:Hide,L:Label;C:Color

- 作图步骤
1) 点击H > lines ,H > spheres,就可隐藏线性结构和球性结构,只保留蛋白螺旋和小分子

2)鼠标点击配体结构,右侧栏中会出现sele,点击A > rename selection 这样我们就可以改名,将sele替换成ligand;
重复上次操作,将sele替换成ligand_res;
在ligand_res这一栏的A > modify > aroud > residues within 4 A ,接着点击S > sticks, 点击H > cartoon ;
就能得到下图配体4埃以内的残基分布、还包含了水分子(加入我们不分析水分子,点击H > waters 就可把水分子隐藏掉):

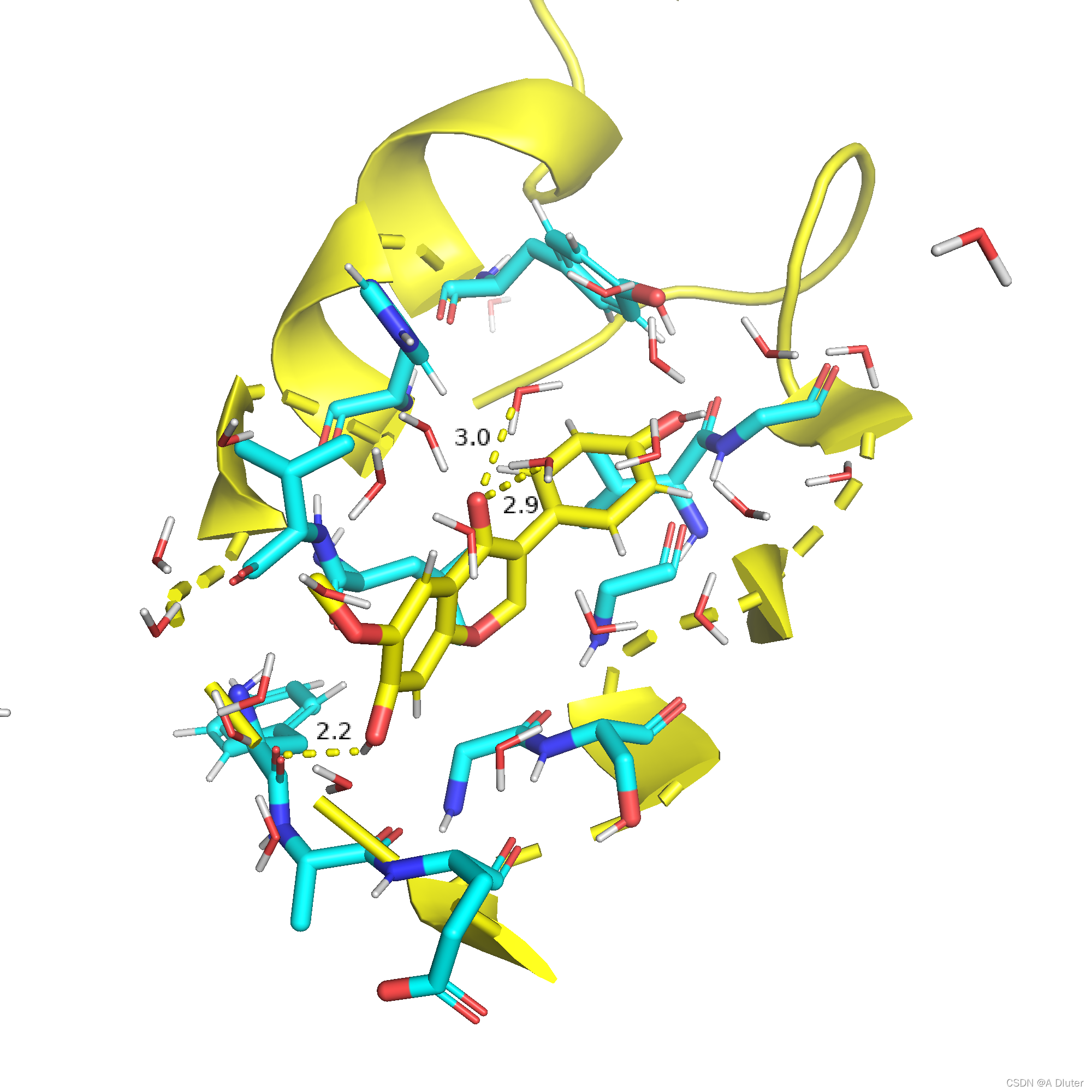
- 接着点击配体,会出现sele一栏,在该栏A > find > polar contacts > to other atoms in object,就会出现黄色的连线,点击sele_polar_conts一栏的S > labels,就会出现氢键距离。
我们可以看到配体和一个残基和两个水分子的氧会生成氢键。

- 命令行指的时红框

输入以下命令就可以渲染出当前的图片
bg_color white #背景颜色调成白色
ray
png hbond.png, 16.93cm, 16.93cm, dpi=300
save hbond.png
- 关于hbond.png所在的位置,我的一般会出现在C:\Users\86183下,如果不知道出现在哪里,就可在电脑中直接搜索hbond.png,这样可以找到
- 做完图,点击File > Save Session as ,这样保存成XXX.pse文件,方便下次修改调整

















 1098
1098

 被折叠的 条评论
为什么被折叠?
被折叠的 条评论
为什么被折叠?
 到【灌水乐园】发言
到【灌水乐园】发言









