






单细胞分析过程遇到的问题
Read10x
利用Read10X这个函数导入数据是真的一波三折
-
首先这个函数是不能导入压缩包形式的文件的,我卡在这里很久,主要也是由于教程太坑人了
如果导入压缩文件,那就会出现下面的报错,也就是找不到文件地址

一定得解压缩,然后就会出现三个文件,这是10x的标准文件(而且这个命名也是很重要的,一定得跟下面这三个一样,后面说)
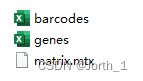
-
文件的命名也很重要
这个数据集解压缩得到的三个文件命名是下面这样,这是10x v3版本
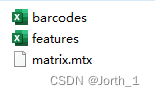
然后就不断出现下面的报错

我就去查了一下,发现是命名的原因

将文档重命名之后就可以运行了
参考文章
Barcode file missing. Expecting barcodes.tsv.gz - 简书 (jianshu.com)
刘小泽学习组合多个单细胞转录组数据 - 简书 (jianshu.com)
Seurat 4 源码解析 4: step1 读入10x数据到内存 Read10X(data.dir=) - 知乎 (zhihu.com)
单细胞分析seurat包学习笔记1 - 简书 (jianshu.com)
sce.all=merge(x=sceList[[1]],
y=sceList[ -1 ],
add.cell.ids = gsub('_gene_cell_exprs_table.txt.gz','',gsub('^GSM[0-9]*_','',samples) ) )
刚开始我看到这句代码时,感觉很奇怪,为什么x一定要选取第一个样品,y就是剩下的样品。其实并不用,只要确保 x 参数是单细胞样品列表中的一个元素即可,可以使用其他任意样品作为 x 参数。也是可以的,最后得到的结果也是一样的。
sce.all_1=merge(x=sceList[[3]], y=sceList[-3])
![[外链图片转存失败,源站可能有防盗链机制,建议将图片保存下来直接上传(img-sVT16JLN-1675524543899)(vx_images/550052821230245.png)]](https://img-blog.csdnimg.cn/1ae1047e5748428fac4215f95543e81e.png)
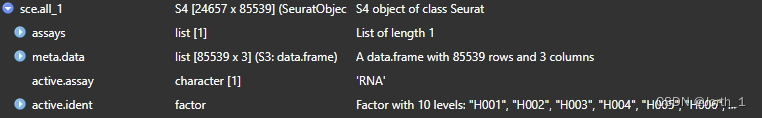
得到的结果是一样,但还是推荐第一个样本作为x,这是为什么呢?接下来我们看下为什么?
> head(sce.all@meta.data, 10)
orig.ident nCount_RNA nFeature_RNA
H001_AAACCCAAGCACTCCG-1 H001 2680 1548
H001_AAACCCACACCGTCGA-1 H001 13725 4266
H001_AAACCCACACGGGTAA-1 H001 5091 2504
H001_AAACCCACAGATCCTA-1 H001 7770 2856
H001_AAACCCACAGCTGGTC-1 H001 1826 1084
H001_AAACCCACAGGTTCCG-1 H001 52893 7531
H001_AAACCCACATGAGTAA-1 H001 23244 6014
H001_AAACCCAGTCCAGAAG-1 H001 4737 1888
H001_AAACCCAGTGCCGTTG-1 H001 12693 4239
H001_AAACCCAGTGTGTGGA-1 H001 4983 2211
行的名字是细胞的名字
> head(sce.all_1@meta.data, 10)
orig.ident nCount_RNA nFeature_RNA
H001_AAACCCAGTATGATCC-1 H003 5567 2502
H001_AAACGAACACAAATGA-1 H003 14882 3995
H001_AAACGAACACGACGAA-1 H003 6351 2322
H001_AAACGAACATGCTGCG-1 H003 20569 4359
H001_AAACGCTAGCAGGTCA-1 H003 12237 3740
H001_AAACGCTCAGGGTTGA-1 H003 9009 2816
H001_AAACGCTCATGCAGCC-1 H003 7864 2726
H001_AAACGCTGTAATTAGG-1 H003 14658 3940
H001_AAACGCTGTGCATGAG-1 H003 13155 3532
H001_AAACGCTTCGCCGATG-1 H003 7674 2614
可以看到这里行的命名是以H001为开头的,为什么呢,这是由于gsub(‘gene_cell_exprs_table.txt.gz’,‘’,gsub('^GSM[0-9]*’,‘’,samples) ),如果想换成H003,需要将代码改一下,但没这个必要,还是从1开始看起来比较好处理。所以还是以第一个样品作为x。至于别的样品可以作为x理解就好了。
add.cell.ids = gsub('_gene_cell_exprs_table.txt.gz','',gsub('^GSM[0-9]*_','',samples) )
这句代码也是需要好好理解一下。那首先需要看下gsub这个函数。
gsub 的第一个参数是需要被替换的字符串,第二个参数是替换字符串的字符(即为空字符串)。第三个参数是需要替换的字符串,即 samples 列表。
它执行了两次字符串替换操作:
第一次:gsub(‘^GSM[0-9]_‘,’',samples),表示删除以 'GSM[0-9]_’ 开头的字符串。
第二次:gsub(‘_gene_cell_exprs_table.txt.gz’,‘’,…),表示删除以 ‘_gene_cell_exprs_table.txt.gz’ 结尾的字符串。
因此,最终结果是删除了列表 samples 中所有元素的指定字符串后的结果。最终结果可以用作单细胞样品的标识符。















 4044
4044











 被折叠的 条评论
为什么被折叠?
被折叠的 条评论
为什么被折叠?
 到【灌水乐园】发言
到【灌水乐园】发言









