









❝Toward the Prediction of Binding Events in Very Flexible, Allosteric, Multidomain Proteins 由 Andrea Basciu 等人撰写,旨在解决灵活多变的多结构域别构蛋白与配体结合预测难题,开发 gEDES 方法并在腺苷酸激酶上验证,对药物设计等领域意义重大。
1. 作者信息
-
Andrea Basciu:意大利卡利亚里大学物理系。研究方向为通过增强采样方法预测蛋白质-配体结合构象,助力药物设计。
-
Alexandre M. J. J. Bonvin:荷兰乌得勒支大学 Bijvoet 生物分子研究中心。专注于生物分子结构与相互作用的计算研究,利用计算方法探究蛋白质-蛋白质、蛋白质-配体相互作用机制。
-
Attilio V. Vargiu:意大利卡利亚里大学物理系。主要研究方向为运用计算生物物理方法,研究蛋白质动态变化及与配体相互作用,为药物设计提供结构基础。
2. 拟解决的问题
在药物发现早期,基于分子对接的虚拟筛选难以模拟蛋白质发生大的构象变化时的配体结合事件,尤其是针对多结构域的别构蛋白。本研究旨在开发一种新方法,仅利用未结合状态的蛋白质结构和假定的结合位点信息,生成高度灵活的别构多结构域蛋白的结合态类似构象,以提高虚拟筛选准确性,助力药物设计
3. 用到的方法
-
gEDES 方法: 基于元动力学模拟,定义多个集体变量(如结合位点的回转半径、跨惯性平面的接触数、准刚性域之间的接触数等),对蛋白质构象进行增强采样,生成结合态类似构象。
-
标准分子动力学模拟: 作为对比方法,采用 AMBER20 软件包中的 pmemd 模块进行标准全原子分子动力学模拟。
-
聚类分析: 对 gEDES 和标准分子动力学模拟产生的轨迹进行聚类分析,在集体变量空间中使用 R 语言的内部脚本,先进行层次聚类,再用 K-均值方法聚类,得到用于对接计算的结构代表。
-
分子对接: 使用 HADDOCK 和 AutoDock 软件,将配体对接至预测的结合位点,评估不同方法生成的蛋白质构象在对接中的表现。
4. 主要发现
-
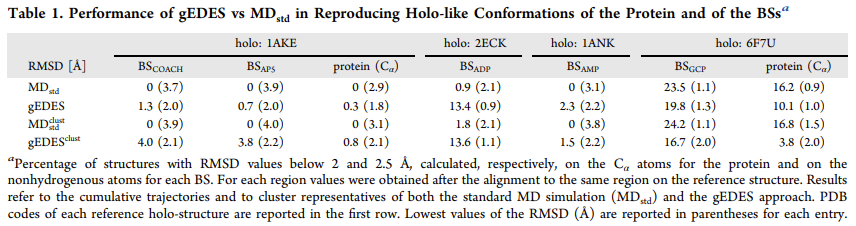
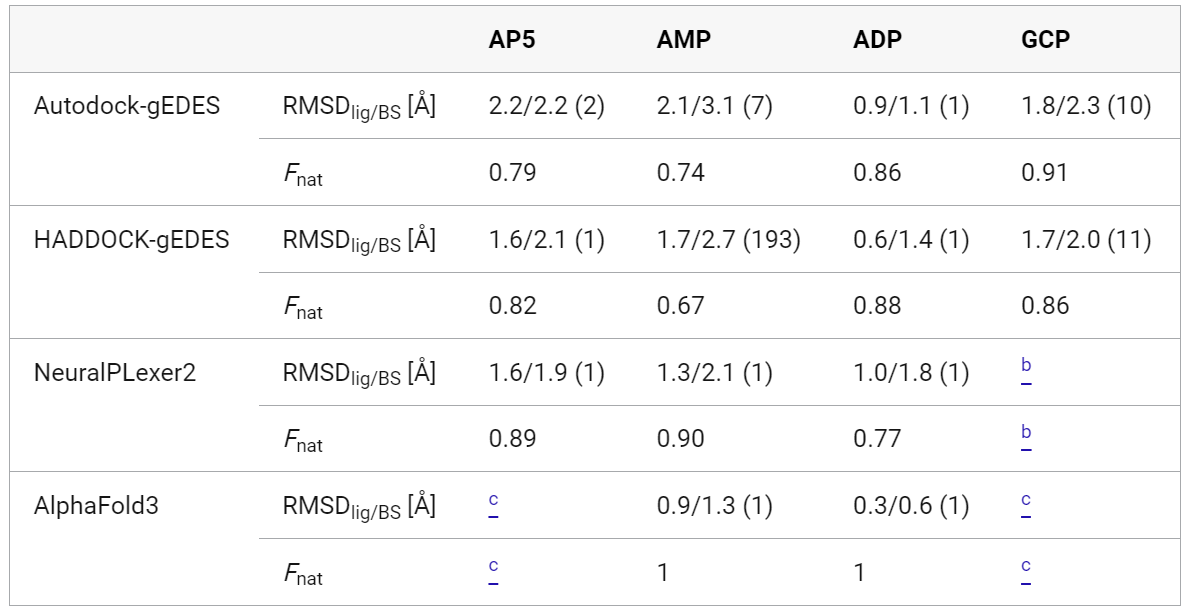
gEDES 能有效生成结合态类似构象: gEDES 可产生大量与多种配体结合态相似的 ADK 结构,在重现结合位点构象方面表现出色,其生成的构象中结合位点的 RMSD 值低至 2.0 Å(整体蛋白为 1.8 Å),而标准分子动力学模拟得到的最佳结构相应 RMSD 值分别为 3.7 Å 和 2.9 Å。
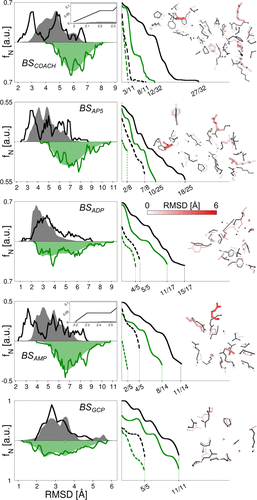
-
gEDES 提高对接准确性: 在对接计算中,使用 gEDES 生成的构象能使多种配体(AP5、ADP、AMP、GCP)的对接结果获得排名靠前的天然样结合模式,而标准分子动力学模拟得到的构象在对接时,即使能得到天然样的配体构象,其排名也较低,且结合位点结构扭曲
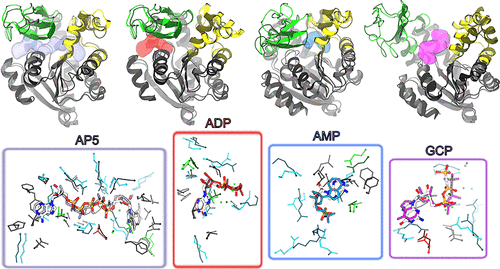
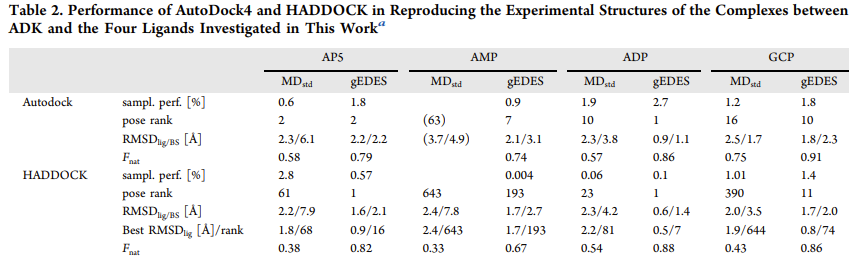
-
gEDES 性能优于其他方法: 与之前的研究方法相比,gEDES 在重现结合位点几何形状方面表现良好,与 NeuralPLexer2 和 AlphaFold3 等方法相比,各有优劣。对于 AP5 和 GCP,NeuralPLexer2 无法准确预测,而 gEDES 能有效预测
总结
-
不足之处: 计算成本较高,模拟时间较长;目前仅在少数蛋白质上进行了验证,其普适性还需进一步在更多不同类型的蛋白质上进行测试;依赖于准确的结合位点预测,若结合位点预测不准确,可能影响最终结果。
-
意义: 为极具挑战性的药物靶点生成结合态类似构象提供了通用框架,可应用于虚拟筛选、预测氨基酸突变对结构和动力学的影响以及蛋白质工程等领域;能准确区分活性和非活性化合物的结合模式和亲和力,对药物设计具有重要意义,有助于发现更多有潜力的先导化合物,或为已上市药物开拓新用途。


















 5307
5307

 被折叠的 条评论
为什么被折叠?
被折叠的 条评论
为什么被折叠?
 到【灌水乐园】发言
到【灌水乐园】发言











