









❝计算蛋白-蛋白、蛋白-DNA、蛋白-小分子体系的结合能(如MM/PBSA或MM/GBSA)是分析复合物结合强度的常用方法。通过将总体结合能分解到各个氨基酸残基上,可以直观地识别哪些残基在结合过程中贡献更大。为了更清晰地展示这些结果,可以在PyMOL中使用渐变色将残基的贡献结合能数值映射到三维结构上。本文将详细介绍如何在PyMOL中实现这一可视化效果。
文中用到的示例文件都可以在同名公主号后台回复:“250221”自取。
接下来,我们以amber分析软件的输出结合能的文件为例进行演示:
-
能量文件名:
FINAL_DECOMP_MMPBSA.dat
蛋白文件名:com1.pdb(该文件已经除去氢原子)
使用如下python脚本,提取FINAL_DECOMP_MMPBSA.dat文件中各个残基的结合能贡献值,并把数值添加到蛋白文件的B因子所在的列。最终输出文件out1.pdb
filename = "FINAL_DECOMP_MMPBSA.dat"
pdbfile = "com1.pdb"#pdb文件需要事先除去氢
residue_list = ["ALA","ARG","ASN","ASP","CYS","GLN","GLU","GLY","HIS","ILE","LEU","LYS","MET","PHE","PRO","SER","THR","TRP","TYR","VAL","CYM","HID","HIP","HIE","CYX" ]
#将单个残基对结合能贡献数值提取到列表
f = open(f'{filename}','r')
i=0
Energy_list = []
for line in f:
i += 1
if i >= 9 and len(line) >= 17:
Energy_list.append(round(float(line.split(",")[17]),2))
#print("Energy(kj/mol):",line.split(",")[17])
#print("std:",line.split(",")[18])
# 将能量数值嵌入b因子所在列
f = open(f'{pdbfile}', 'r')
line_list = []
new_line_list = []
i = 0
j = 0
for line in f:
if len(line.split()) >= 3:
i += 1
if i < 2:
line_list.append(line)
new_line = '{0:60}{1:>6}'.format(line[0:60], Energy_list[j])
new_line_list.append(new_line)
if i >= 2:
line_list.append(line)
resname = line[17:20]
resid = line[20:26]
if resid == line_list[i-2][20:26] and resname in residue_list:
new_line = '{0:60}{1:>6}'.format(line[0:60], Energy_list[j])
new_line_list.append(new_line)
elif resname in residue_list:
j += 1
new_line = '{0:60}{1:>6}'.format(line[0:60], Energy_list[j])
new_line_list.append(new_line)
print("最小贡献残基能量:",max(Energy_list))
print("最大贡献残基能量:",min(Energy_list))
#输出文件
with open("out.pdb", 'w') as f1:
for line in new_line_list:
f1.write(line + '\n')
将上述内容存储为run.py,执行改脚本文件:
python run.py
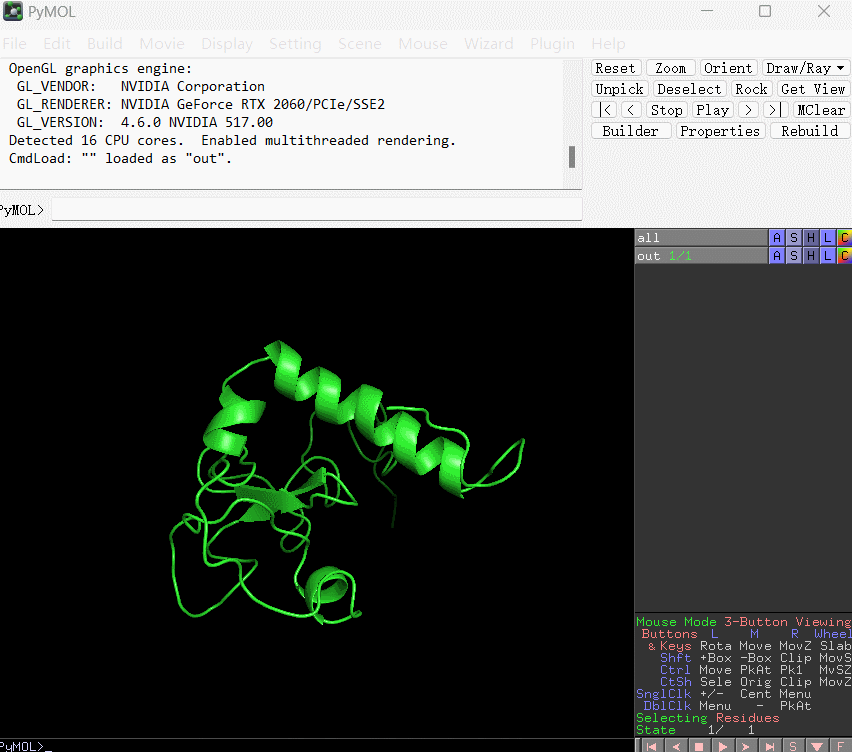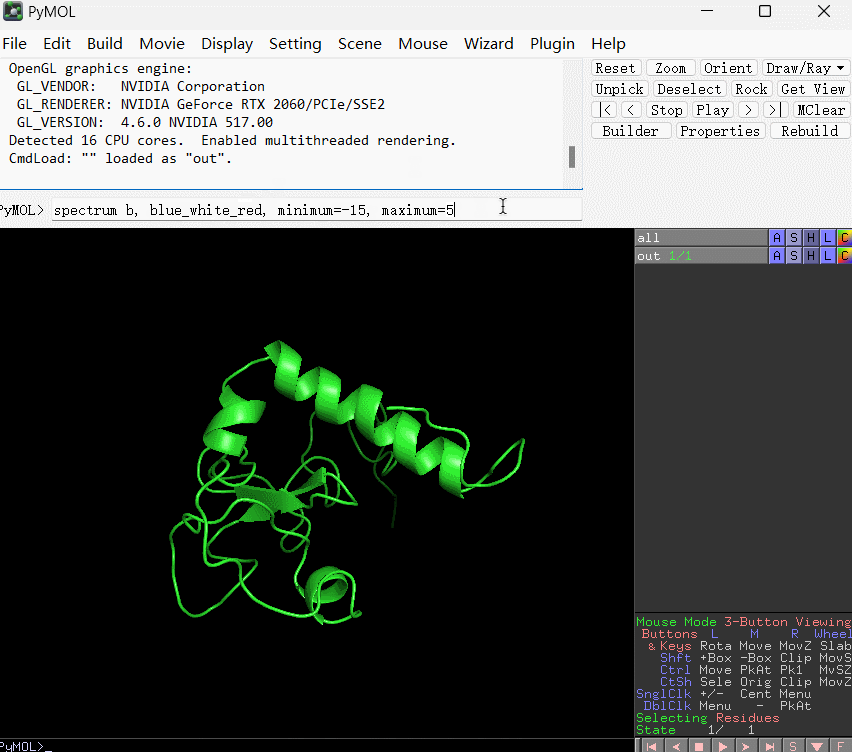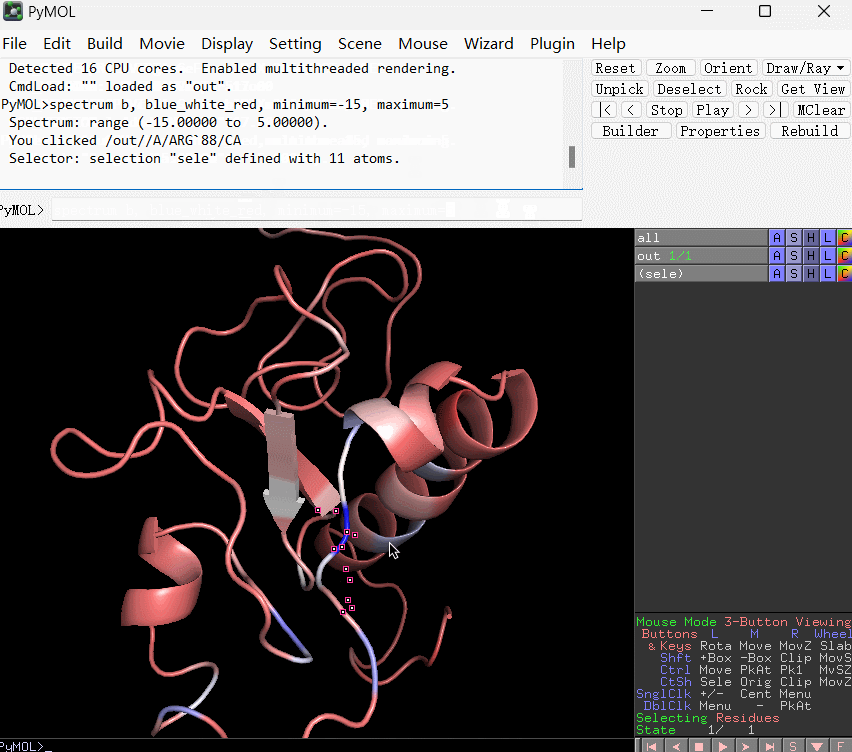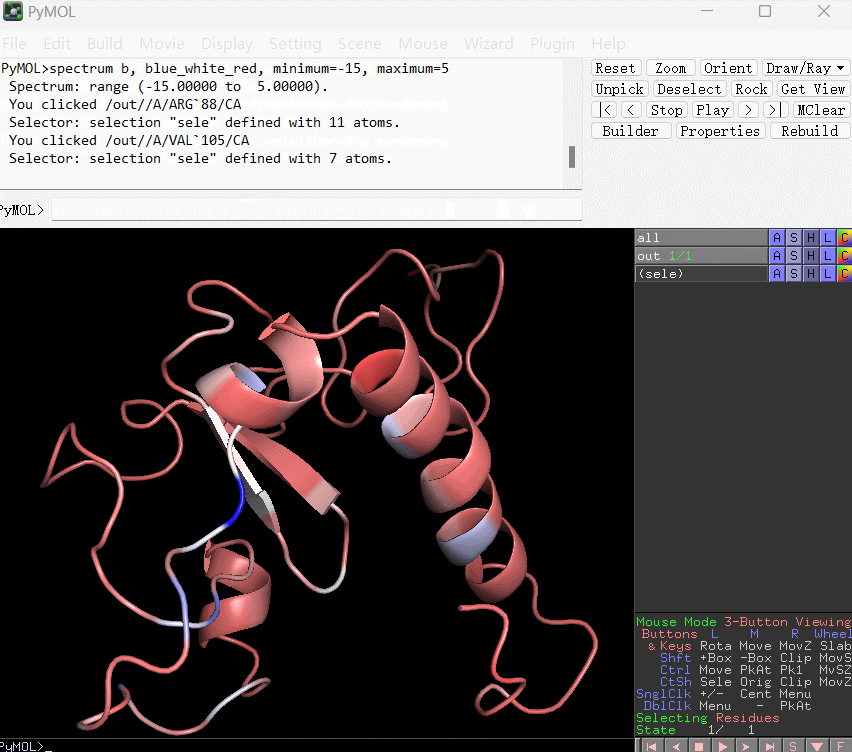
此时你会得到文件out.pdb,使用pymol打开,用b-factor的着色方式显示(如下图所示):
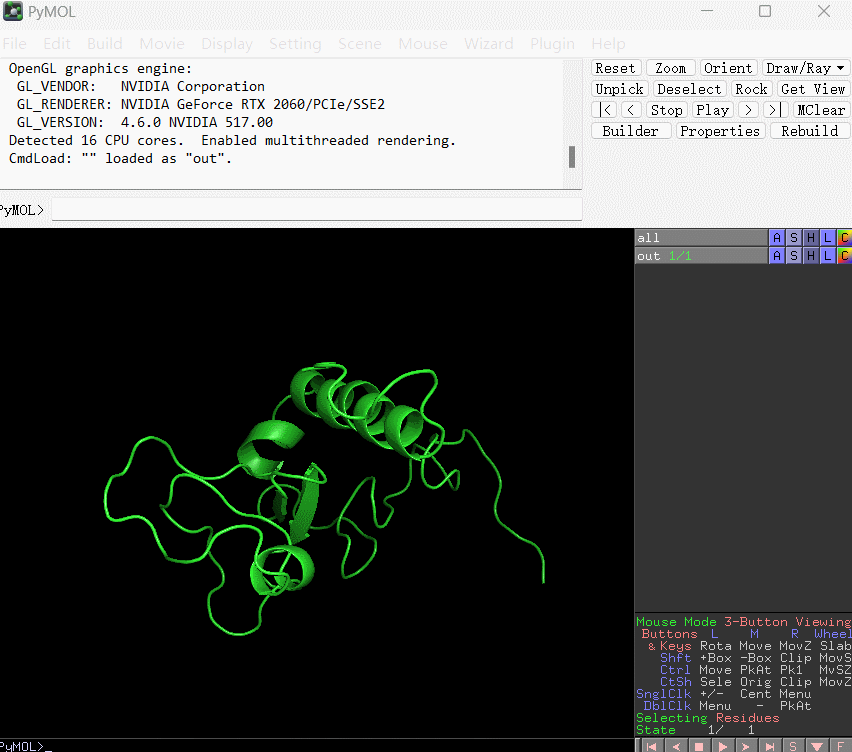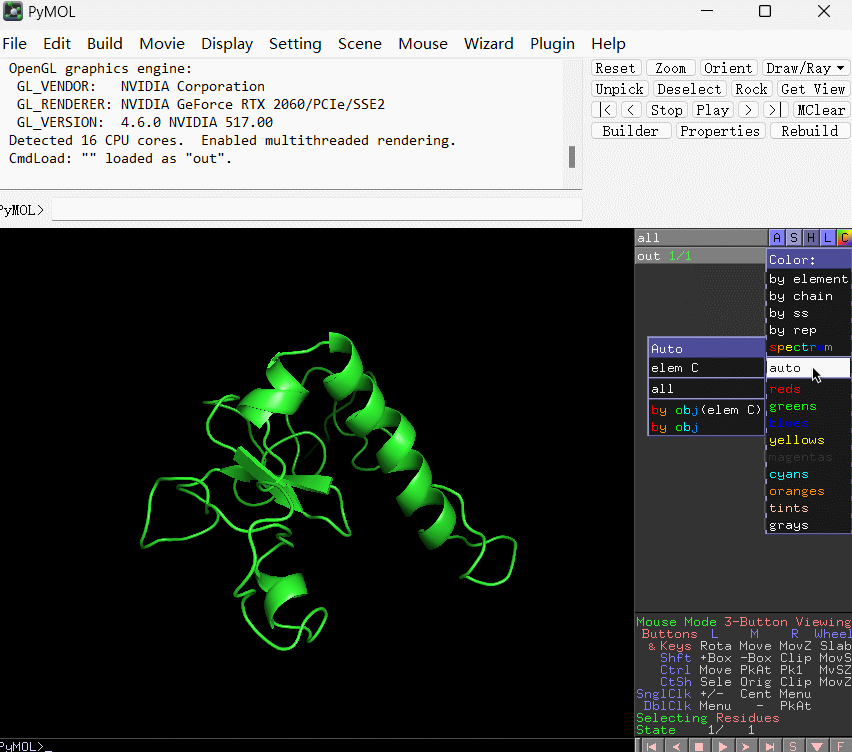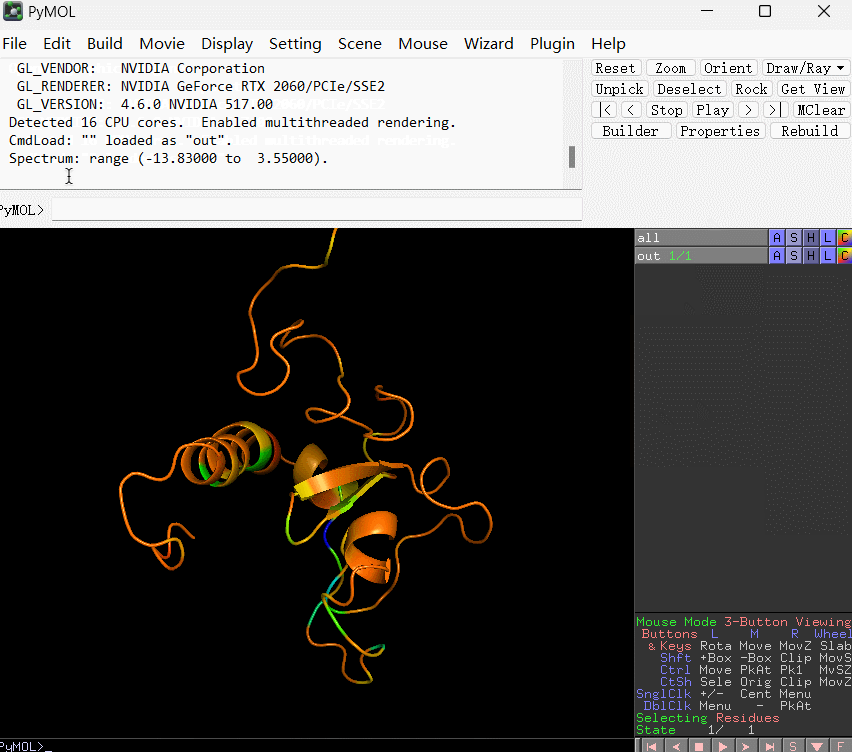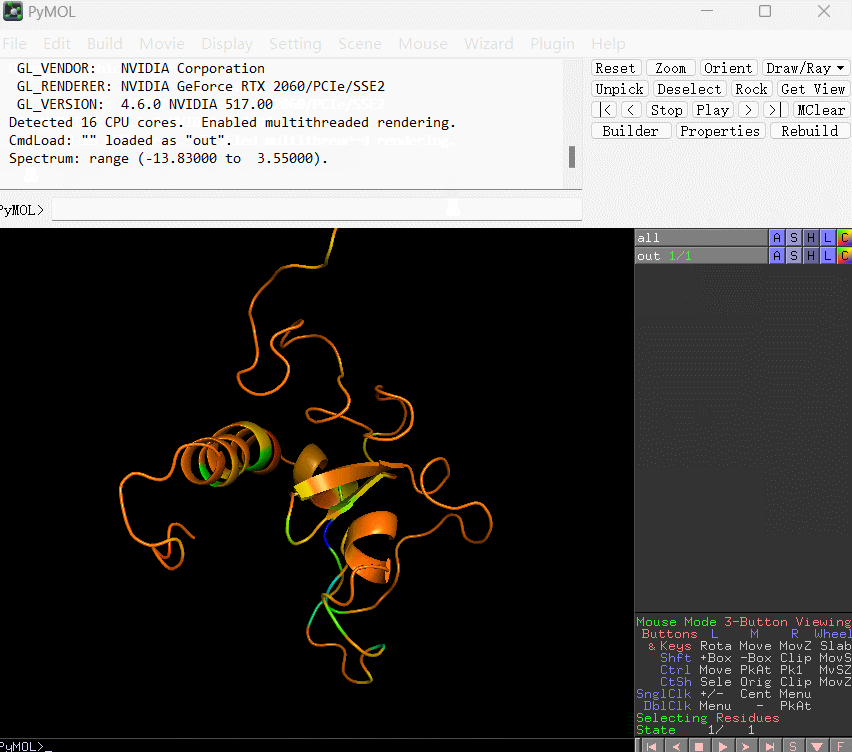
默认颜色越偏向暖色调表示残基的结合能数值越是偏大,相应地对结合的贡献越小;反之颜色越是偏向冷色调表明该残基的结合能越小,相应地对结合贡献越大。
当然,如果你想要在pymol中自定义着色方式,以及自定义颜色和数值的对应范围,可以在pymol内部命令行内输入下条语句:
spectrum b, blue_white_red, minimum=-15, maximum=5
上条语句的含义是:将 B-factor(这里替换成了残基的能量贡献数值) 在 -15 到 5 之间映射为从蓝到白再到红的渐变。使用演示如下所示:



















 3579
3579

 被折叠的 条评论
为什么被折叠?
被折叠的 条评论
为什么被折叠?
 到【灌水乐园】发言
到【灌水乐园】发言











