









细胞器基因组核酸多样性(pi)计算
在分析细胞器基因组时,如叶绿体基因组、动物线粒体基因组,常常会计算多个序列之间的核酸多样性(pi)。步骤一般如下:
-
准备好原始序列
-
调整序列的方向和起点
-
使用比对软件将序列对齐
-
计算pi值
那么问题来了,pi值反应了什么?pi值和序列的相似度有什么联系?为什么常用的如dnasp软件计算的结果中,窗口位置和中点位置不是自己设置的值?其他工具,如vcftools等,计算的pi值和dnasp计算的值为什么不等?
pi值反应什么,要先了解pi是怎么计算的。可以简单的理解成一个概率问题:在对齐后的序列中,从某个区域相同的位点上(排除有空位比对的位点),抽取两个碱基,这两个碱基不相同的平均概率即为pi值。如下图:

使用滑窗计算的时候,带有空位的点也是不参与到pi计算中的。如AAATTTATTTT、AAATT-ATTTT,对齐后的长度是11 bp,但是在计算整体pi的时候,总长度是10 bp。如果设置窗口是5 bp,步长也为5 bp的话,第一个窗口为:1-5,中点为:3;第二个窗口为:6-11,中点为:9。因为第一个窗口中(1-5)没有空位,位置正常。第二个窗口中第一个位置是空位,不计入窗口大小,所以是6-11(其实算的是7-11,如果中间还有空位,则会继续向后跳过),中点也是去掉空位后计算得到的中点(但是最终会映射到对齐后序列中的位置),不是(6+11)/2。多序列中含有一个空位就会跳过该位置,所以对齐后的序列空位越多,窗口显示的实际长度越长(超过设置的大小)。
此外,在刚才的序列中,pi值为0,但是相似度不是100%,因为序列有空位。所以在有些同源性分析的软件中,例如mvista,某些区域的相似度可能比较低,但是pi值也很小,从而造成两者结果不匹配的假象,这种情况可能就是因为该区域空位比对较多。
因此pi实际上反应的是序列中snp(单核苷酸多态性)的多态性,不包含存在任何indel(插入、缺失)的位点。snp的信息可以保存在vcf格式的文件中,所以也可以直接对vcf文件进行pi值计算(有的工具尽管输入的是对齐后的序列,但仍然会转换成vcf格式的文件进行计算,如:perl中的Bio::PopGen::Statistics模块)。但是软件进行计算的时候,会把每个样品变成两条序列(单倍型的序列只对应0/0、或者1/1类型),如上面的例子,直接用序列计算的时候是两条,但是如果转换成vcf格式再进行计算时,实际就成了四条如AAATTTATTTT/AAATTTATTTT、AAATT-ATTTT/AAATT-ATTTT。所以计算出来的值就有些差异,vcf文件计算的pi值会稍微变大一些(变成原来的2*(n-1)/(2n-1),n为样品的个数),不过该例子中pi为0,不影响。所以在对单倍型序列进行pi值计算的时候,最好直接选择dnasp这种对原始序列进行计算的软件。
下面是我自己编写的python脚本,供参考。
# 运行方式:
python3 calculate_pi.py aln.fa window.size step > pi.stat.xls
#!/usr/bin/env python3
# modified on 2023.07.24
# author:awk_bioinfo
import sys
from itertools import combinations
align_file = sys.argv[1]
window = int(sys.argv[2])
step = int(sys.argv[3])
def Cab(m,n):
a=b=result=1
if m<n:
print("n不能小于m 且均为整数")
elif ((type(m)!=int)or(type(n)!=int)):
print("n不能小于m 且均为整数")
else:
minNI=min(n,m-n)#使运算最简便
for j in range(0,minNI):
#使用变量a,b 让所用的分母相乘后除以所有的分子
a=a*(m-j)
b=b*(minNI-j)
result=a//b #在此使用“/”和“//”均可,因为a除以b为整数
return result
def generPiDict(align_file):
alnDict = {}
PiDict = {}
seqnum = 0
with open(align_file,'r') as fp:
#site = 0
for line in fp:
if line.startswith('>'):
site = 0
seqnum += 1
else:
LN = line.strip()
alnlength = len(LN)
#print(alnlength)
for i in range(alnlength):
alnDict.setdefault(site,[]).append(LN[i])
site +=1
for k,sList in alnDict.items():
if '-' in sList:
PiDict[k] = -1
else:
result = combinations(sList, 2)
cnt=0
for s in result:
if s[0] != s[1]:
cnt += 1
# [cnt = cnt + 1 for s in result if s[0] != s[1]] ?
PiDict[k] = cnt
return PiDict, seqnum
PiDict, seqnum = generPiDict(align_file)
wholelength = len(PiDict.keys())
cab = Cab(seqnum,2)
########################### 计算Pi值 ######################
##### 1.配置参数 #####
start = 1
spi = []
extendWindow = window
midwindow = int(window/2)
extendStep = step
j=0
k=0
l=0
site=0
mid=0
#extendStep2 = ''
##### 配置参数 #####
#打印参数和表头信息
print(f'''
#seq number: {seqnum}
#aln length: {wholelength}
#window length: {window}
#step size: {step}
#===================================
'''.strip()
)
print('Window', 'Midpoint', 'Pi', 'S', sep="\t")
###### 2.计算逻辑 #####
#注意: 返回实际步长后又得跳回去重新计算
while start < wholelength:
for i in range(start-1,wholelength):
PiStat = PiDict[i]
#print(PiStat)
if PiStat == -1:
extendWindow +=1
extendStep +=1
midwindow +=1
else:
spi.append(PiStat)
j +=1
k += 1
l += 1
if PiStat > 0:
site += 1
if k == step:
extendStep2 = extendStep # 窗口取最长值
if l == int(window/2): ## 注意此处:如果下一个是-1,则l不变,但midwindow会+1,需取第一次Midpoint值
if not mid:
mid = start + midwindow - 1
if j == window:
print(f"{start}-{start + extendWindow - 1}", mid, round(sum(spi)/(cab * window),5), site, sep="\t")
start = start + extendStep2
spi = []
extendWindow = window
midwindow = int(window/2)
extendStep = step
j=0
k=0
l=0
site=0
mid=0
break
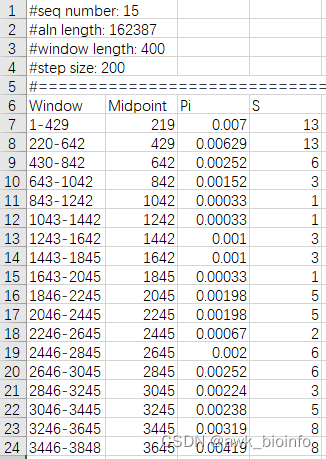
结果展示:
DnaSP的结果:
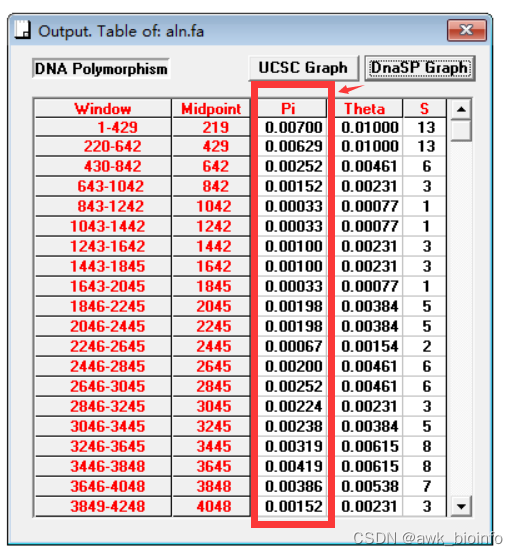
有同学对脚本计算结果提出了质疑。小编重新构思,连夜对代码做了更新。对比了经典软件DnaSP计算核苷酸多样性的结果,窗口和Pi值均相同。供参考。
参考:https://mp.weixin.qq.com/s/4BSMhd2zm-s2NUJh3aZLIw


















 790
790

 被折叠的 条评论
为什么被折叠?
被折叠的 条评论
为什么被折叠?
 到【灌水乐园】发言
到【灌水乐园】发言









