







library(ggplot2)
library(magrittr)
library(dplyr)
library(stringr)
options(stringsAsFactors = F)
options(as.is = T)
setwd('~/Desktop/kk_result20201101/')
导入数据
#df1 = read.table("GSE116174kk_down.txt",sep = '\t',header = T)
#df2 = read.table("GSE116174kk_up.txt",sep = '\t',header = T)
df3 = read.table("GSE14520kk_down.txt",sep = '\t',header = T)
df4 = read.table("GSE14520kk_up.txt",sep = '\t',header = T)
df5 = read.table("ICGCkk_down.txt",sep = '\t',header = T)
df6 = read.table("ICGCkk_up.txt",sep = '\t',header = T)
df7 = read.table("TCGAkk_down.txt",sep = '\t',header = T)
df8 = read.table("TCGAkk_up.txt",sep = '\t',header = T)
#df1$dataset = '6174dn'
#df2$dataset = '6174up'
df3$dataset = '14520dn'
df4$dataset = '14520up'
df5$dataset = 'icgcdn'
df6$dataset = 'icgcup'
df7$dataset = 'tcgadn'
df8$dataset = 'tcgaup'
分别提取上下调基因
合并数据,如果只有一套数据的话不要执行这个步骤
dfdn = NULL
#dfdn = rbind(dfdn,df1)
dfdn = rbind(dfdn,df3)
dfdn = rbind(dfdn,df5)
dfdn = rbind(dfdn,df7)
dfup = NULL
#dfup = rbind(dfup,df2)
dfup = rbind(dfup,df4)
dfup = rbind(dfup,df6)
dfup = rbind(dfup,df8)
筛选 FDR
fdr=0.05
dfdn = dfdn[which(dfdn$FDR<fdr),]
dfdn = dfdn[which(dfdn$Term %in% names(which(table(dfdn$Term) > 1))),]
dfdn$y = str_split(dfdn$Term,':',simplify = T)[,2]
dfdn$x = log10(dfdn$FDR)
dfup = dfup[which(dfup$FDR<fdr),]
dfup = dfup[which(dfup$Term %in% names(which(table(dfup$Term) > 1))),]
dfup$y = str_split(dfup$Term,':',simplify = T)[,2]
dfup$x = -log10(dfup$FDR)
dfdn$dir = 'dn'
dfup$dir = 'up'
将上下调数据合并
df = rbind(dfdn, dfup) %>% as.data.frame()
df$dataset1 = str_split(df$dataset,'up|dn',simplify = T)[,1]
label1 = df[order(df$dataset1,df$x),]
label1 = label1$y
label1 = label1[!duplicated(label1)]
ggplot(data=df,aes(x,y,fill =dir ))+
scale_y_discrete(limits=label1)+
geom_bar(stat="identity", color="black", position = 'dodge')+
scale_fill_manual(values=c("#00AFBB", "#FC4E07"))+theme_bw()+
facet_wrap(~dataset1)
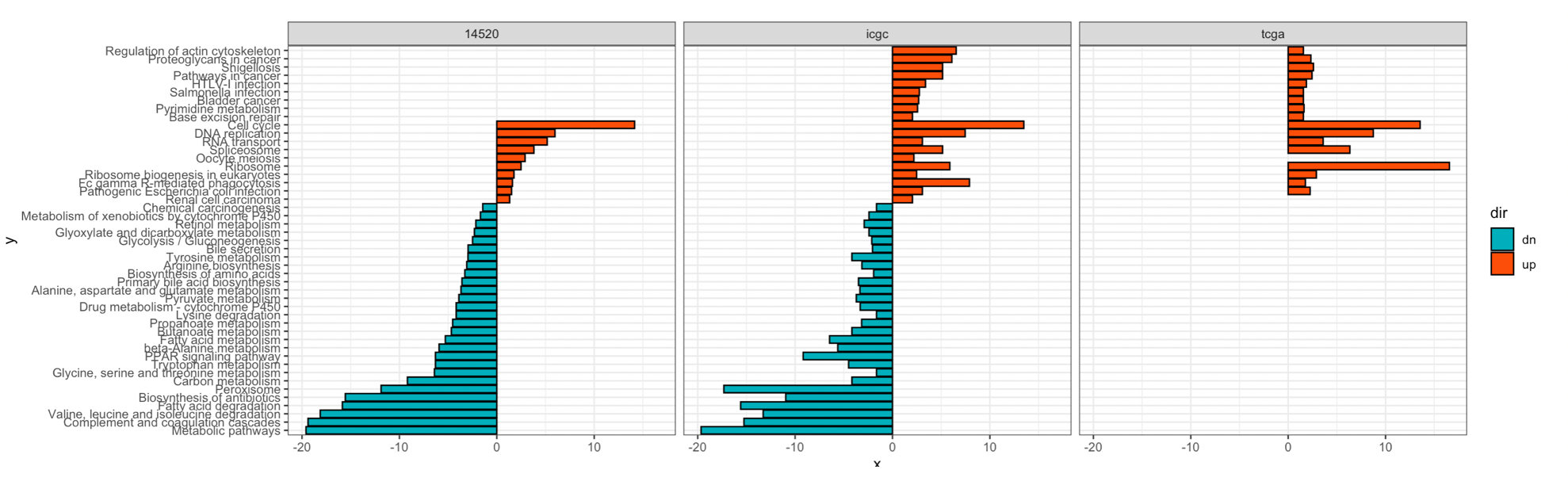
转载内容
差异分析的结果**
这个结果实际上已经通过counts函数获得了,我们不在担心,处理组和对照组完全相反这种情况,因为他内置了参数设定比较组。比如,我们有5个处理组,我们不需要做5次Deseq,我们在results中指定即可。
用summary这个函数,可以看到差异分析的结果,高表达和低表达的比例。低丰度基因所占的比例。
summary(dd2, alpha = 0.05)

再把差异分析的结果转化成data.frame的格式
library(dplyr)
library(tibble)
res <- dd2 %>%
data.frame() %>%
rownames_to_column("gene_id")
基因ID转换
以前我们从gtf文件转换的, 但是我们需要gtf文件来提取mRNA以及lncRNA,就顺手做了ID转换,而且,mRNA和lncRNA是分别做的Deseq2,这从原理上来讲,是有问题的,Deseq2矫正了测序的深度,而这个深度应该是所有基因算在一起的深度,不应该分开来算。
TGCA数据的标准化以及差异分析
用两个包来转换,得到ENTREZID用于后续分析,得到SYMBOL便于识别。
library(AnnotationDbi)
library(org.Hs.eg.db)
res$symbol <- mapIds(org.Hs.eg.db,
keys=res$gene_id,
column="SYMBOL",
keytype="ENSEMBL",
multiVals="first")
res$entrez <- mapIds(org.Hs.eg.db,
keys=res$gene_id,
column="ENTREZID",
keytype="ENSEMBL",
multiVals="first")
有了这个文件,里面有logFC,p值,还有基因名称,我们可以完成GO,KEGG,热图,火山图所有操作。这一部分内容参考
最有诚意的GEO数据库教程
这个帖子目前已经有接近4000次访问,这在我们这样一个小号是不容易的,靠的是真诚。
制作geneList
我们在那个帖子里面并没有讲GSEA分析,今天来展示一下。原理略过。我们这里还是用Y叔的神包clusterprofier,神包虽好,记得引用。
使用这个包做GSEA,要制作一个genelist,这个是一个向量,他的内容是排序后的logFC值,他的名称是ENTREZID,而这两个我们都是不缺的,在上一步得到的差异结果中。
library(dplyr)
gene_df <- res %>%
dplyr::select(gene_id,log2FoldChange,symbol,entrez) %>%
## 去掉NA
filter(entrez!="NA") %>%
## 去掉重复
distinct(entrez,.keep_all = T)
制作genelist的三部曲
## 1.获取基因logFC
geneList <- gene_df$log2FoldChange
## 2.命名
names(geneList) = gene_df$entrez
## 3.排序很重要
geneList = sort(geneList, decreasing = TRUE)
看一下这个genelist,增加感性的理解
head(geneList)

GSEA分析
完成KEGG的GSEA分析
library(clusterProfiler)
## 没有富集到任何数据
gseaKEGG <- gseKEGG(geneList = geneList,
organism = 'hsa',
nPerm = 1000,
minGSSize = 20,
pvalueCutoff = 0.05,
verbose = FALSE)
作图展示富集分布图
library(ggplot2)
dotplot(gseaKEGG,showCategory=12,split=".sign")+facet_grid(~.sign)
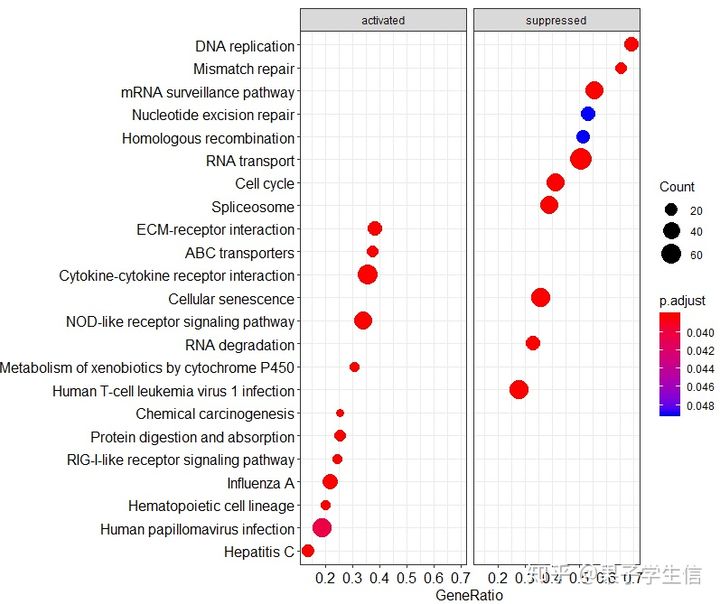
这时候,我们看到有一些通路是被激活的,有一些通路是被抑制的。比如Cell cycle是被抑制的,我们可以选取单个通路来作图。
把富集的结果转换成data.frame,找到Cell cycle的通路ID是hsa04110
gseaKEGG_results <- gseaKEGG@result
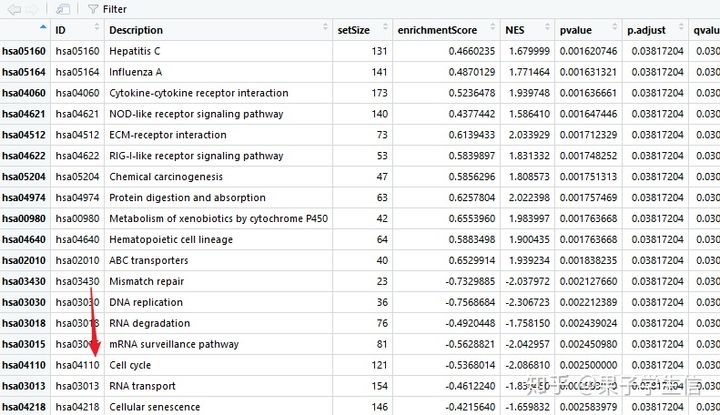
使用gseaplot2把他画出来
library(enrichplot)
pathway.id = "hsa04110"
gseaplot2(gseaKEGG,
color = "red",
geneSetID = pathway.id,
pvalue_table = T)
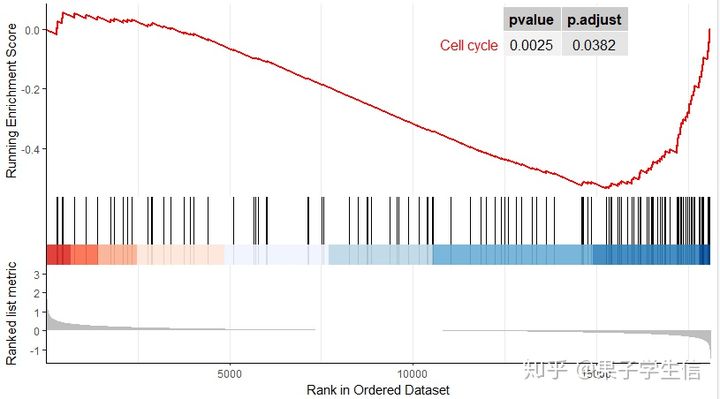
也可以画出一个激活的
pathway.id = "hsa04060"
gseaplot2(gseaKEGG,
color = "red",
geneSetID = pathway.id,
pvalue_table = T)
pathview 展示
我们现在知道cell cycle是被抑制的,如果还想看一下这个通路里面的基因是如何变化的,应该怎么办呢,pathview 可以帮到我们。
library(pathview)
pathway.id = "hsa04110"
pv.out <- pathview(gene.data = geneList,
pathway.id = pathway.id,
species = "hsa")
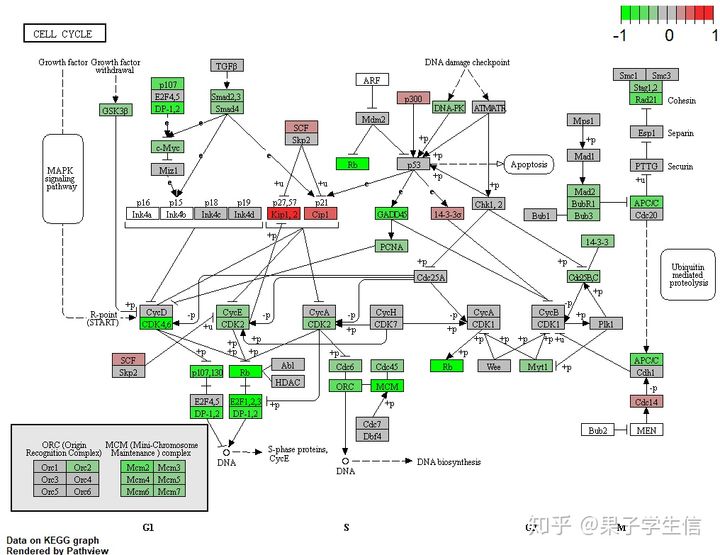
改变一下参数,可以得到另外一种构图
library(pathview)
pathway.id = "hsa04110"
pv.out <- pathview(gene.data = geneList,
pathway.id = pathway.id,
species = "hsa",
kegg.native = F)

一眼看过去,都是绿的,说明这个通路确实是被抑制了,还可以在图上缕一缕,哪些是核心分子,一般说来,越往上游越核心。















 837
837











 被折叠的 条评论
为什么被折叠?
被折叠的 条评论
为什么被折叠?
 到【灌水乐园】发言
到【灌水乐园】发言









